




Investigation of novel SARS-COV-2 variant
Variant of Concern 202012/01
Detection of an epidemiological cluster associated with a new variant of concern
Analysis of routinely available genomic data in Kent, England was undertaken as part of an epidemiological investigation to investigate increasing incidence on 8 December 2020. Although only 4% (255/6130) of Kent cases had available genomes through COG-UK sequencing, a large phylogenetic luster of 117 genomically similar cases over the week 10-18 November 2020 was identified.
The Kent cluster, when examined in the national phylogeny, is part of a larger cluster (962 genomes at the time of analysis on 8 December 2020). This cluster is phylogenetically very distinct from the rest of the UK dataset. These cases were concentrated in Kent and NE London, with limited spread into the rest of London, Anglia and Essex.
Of the 962 cases in the cluster, data was available for 915 individuals; most specimen dates were in November (828/915) followed by October (79/915), with a small number of cases in September (4/915). Distribution of cases by patient sex is similar (51% female, 49% male). By age, just under 90% of individuals are aged <60 years; work is being undertaken to compare this age distribution to relevant comparators. Six of the 915 cases are deceased.
Nomenclature of variants in the UK
SARS-COV-2 variants if considered to have concerning epidemiological, immunological or pathogenic properties are raised for formal nvestigation. At this point they are designated Variant Under Investigation (VUI) with a year, month, and number. Following risk assessment with the relevant expert committee, they are designated Variant of Concern (VOC). This variant was designated VUI-202012/01 on detection and on review re-designated as VOC-202012/01 on 18/12/20.
Current epidemiological findings
The cluster has spread geographically. As of 20 December 2020, the regions in England with the largest number of confirmed cases with the variant are London, South East and East of England regions.
The UK has a high throughput national testing system for community cases based in a small number of large laboratories. Three of these laboratories use a three target assay (N, ORF1ab, S) from Thermo Fisher (TaqPath). Currently more than 97% of pillar 2 PCR tests which test negative on the S-gene target and positive on other targets are due to the VOC (cf. Section Impact on diagnostic assay below).
We therefore use the frequency of S-gene target negatives among PCR positives as a proxy for frequency of the VOC. This proxy has a limited time window, and is generally a poor proxy the further back in time considered due to other older virus variants which also test negative on spike. We consider the period of calendar weeks 44-49 (25 October-5 December). Examining the proportion of pillar 2 PCR tests negative and positive for the S gene target and stratifying by STP and week, we calculate the week on week growth rate in both S-negative and S-positive cases by simply dividing the case numbers in week t+1 by the case numbers in week t. We correct these weekly growth factors by raising them to the power of 6.57 to ensure they can be interpreted as reproduction numbers (given the mean generation time of SARS-CoV-2). For each STP and week, we compute the ratio of the resulting empirical reproduction number of the S-negative cases to that of the S-positive cases. This yields a measure of the transmission fitness of the variant relative to pre-existing strains. Figure 1 shows the mean ratio of growth factors (corrected by power of 6.5/7 to give reproduction number scale) = 1.47 (95% CI: 1.34-1.59).
Frequency of the VOC was associated with estimated epidemic growth rates. Statistical modelling of the relationship between the frequency of VOC detection and the reproduction number Rt indicates that the VOC is significantly associated with a higher reproduction number. We find that Rt increases by 0.57 [95%CI: 0.25-1.25] when we use a fixed effect model for each area. Using a random effect model for each area gives an estimated additive effect of 0.74 [95%CI: 0.44- 1.29]. This is based on 1419 VOC genomes and 33,792 non-VOC genomes collected in weeks 42 to 48 and with results aggregated weekly at the level of NHS STP regions. The estimates for Rt were obtained using COVID-19 pillar 2 PCR-positive case counts and deaths using a previously developed Bayesian semi-mechanistic transmission model with a latent weekly random walk process that makes no assumptions about the underlying factors driving transmission.
Spike gene target failure (SGTF) can serve as a proxy for carriage of the VOC (cf. Section Impact on diagnostic assay below). Classifications of SGTF are preliminary as case definitions are still being developed. On this basis, we adjusted rates of SGTF for variable specificity over time and between local authorities and then applied the same models to estimate the association of VOC frequency and reproduction number. This analysis shows an increase of Rt of 0.52 [95%CI: 0.39-0.70] when we use a fixed effect model for each area. We also fitted a similar model but with a random effect model on the area, giving an estimated additive effect of 0.60 [95%CI:0.48 - 0.73]. Similar estimates of 0.56 [95%CrI:0.37-0.75] were obtained using a Bayesian regression model accounting for errors in VUI frequencies and Rt estimates. As an example, under the fixed effect model, an area with an Rt of 0.8 without the new variant would have an Rt of 1.32 [95%CI:1.19-1.50] if only the VOC was present.
Among 40 local authorities in East and South East England with more than five VOC samples there is a significant trend of increasing reported cases with increasing frequency of N501Y (Figure 1, weighted linear regression p=10-6). A 10% difference in VOC frequency in mid-November corresponds to approximately 50 more weekly cases per 100 thousand in early December. Local authorities with few VOCsamples have similar reported cases as the rest of the UK (linear regression intercept = 137 cases per 100k versus UK median 130.4 per 100k).
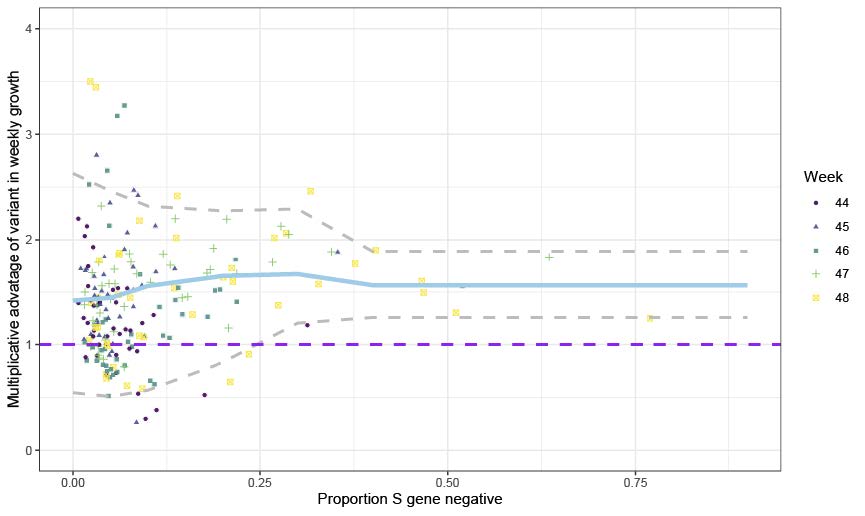
Figure 1. Empirical data analysis of the multiplicative advantage in weekly growth rates. Each point represents the ratio of weekly growth rates between VOC and non-VOC for an NHS England STP area and week, based on the pillar 2 data shown in Figure S1. Colours and shapes differentiate calendar weeks. Numbers above 1 show a multiplicative advantage. The blue line represents the mean value for a particular frequency, and the grey lines the 95% envelope. Scatter at low frequencies largely reflects statistical noise due to low counts.
Genomic characteristics of the VOC
The new variant is defined by 23 mutations: 13-non synonymous mutations, 4 deletions and 6 synonymous mutations. The non-synonymous mutations include a series of spike protein mutations (Table 1). Other notable mutations include a stop codon in ORF8. There are 6 synonymous mutations with 5 in ORF1ab (C913T, C5986T, C14676T, C15279T, C16176T), and one in the M gene (T26801C). This is an unusually large number of mutations in a single cluster.
Lineage-defining protein altering mutations defining the new variant.
| Gene | Nucleotide | Amino acid | |
| ORF1ab | C3267T | T1001I | |
| C5388A | A1708D | ||
| T6954C | I2230T | ||
| 11288-11296 deletion | SGF 3675-3677 deletion | ||
| spike | 21765-21770 deletion | HV 69-70 deletion (Click to more details about HV 69-70 deletion related products) | |
| 21991-21993 deletion | Y144 deletion | ||
| A23063T | N501Y (Click to more details about N501Y related products) | ||
| C23271A | A570D | ||
| C23604A | P681H | ||
| C23709T | T716I | ||
| T24506G | S982A | ||
| G24914C | D1118H | ||
| Orf8 | C27972T | Q27stop | |
| G28048T | R52I | ||
| A28111G | Y73C | ||
| N | 28280 GAT->CTA | D3L | |
| C28977T | S235F |
B.1.1.7 has an unusually large number of genetic changes, particularly in the spike protein. Three of these mutations have potential biological effects that have been described previously to varying extents:
• Mutation N501Y is one of six key contact residues within the receptor-binding domain (RBD) and has been identified as increasing binding affinity to human ACE2.
• The spike deletion 69-70del has also occurred a number of times in association with other RBD changes.
• Mutation P681H is immediately adjacent to the furin cleavage site, a known location of biological significance.
The most unusual and concerning single mutation in this cluster is N501Y. However, the summative effect of this large number of mutations is also unknown and of concern.
Similar to Danish Cluster 5, it may suggest that the virus has replicated under different selective pressures, for example in an alternative host or possibly in an immunocompromised patient, although this is speculative. A recent case report of an immunocompromised individual persistently infected with SARS- COV-2 acquired approximately 10 mutations in the spike protein over 154 days, notably including N501Y. Further discussion can be found at: https://www.genemedi.net/i/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations
Potential impact of spike variant N501Y
Transmissibility: It is highly likely that N501Y affects the receptor binding affinity of the spike protein and it is possible that this mutation alone or in combination with the deletion at 69/70 in the N terminal domain (NTD) is enhancing the transmissibility of the virus. This is based on the position of the 501 residue in the spike receptor binding domain and data showing that N501Y increases spike interactions with human ACE2. N501Y is one of a number of artificially generated RBD variants shown to do this (others include Y453F and N439K). It should be noted that this mutation is the only spike variant found to date in mouse-adapted SARS-CoV2 and is also seen in ferret infections.
Antigenicity: Position 501 is in the RBD, where neutralising antibodies most frequently act, and therefore it is possible that variants at this position affect the efficacy of neutralisation of virus. Of several monoclonal antibodies tested across different studies, one (LYCoV016) showed decreased ability to neutralise SARS-CoV2 variants with mutations at position 501. N501Y was not included. There is currently no neutralisation data on N501Y available from polyclonal sera from natural infection.
Other spike variants
Much less is known about the other spike variants present in this cluster, with the exception of D614G which is well characterised and already highly prevalent in the UK. Their significance cannot be judged at present. Deletion at position 69/70 was present in the Danish Cluster 5 and has been seen in other clusters. Its significance is unknown. Deletions in the 145 area have been noted in infections in immunocompromised patients. Residues at positions 570, 681, 716, 982 and 1118 are of unclear significance although they fall in potentially structurally important areas of the spike protein.
There is a small amount of data about variants affecting ORF8, a viral accessory protein which may be involved in immune evasion by downregulation of MHC class I. In Singapore an ORF8 deletion was associated with attenuated disease, but this was not supported by findings in primary human airway cell experiments.
Impact on diagnostic assays
The UK has a high throughput national testing system for community cases based in a small number of large laboratories. Three of these laboratories use a three-target assay (N, ORF1ab, S). S gene target failures (in otherwise positive samples) began to increase dramatically from late November.
The VOC includes a deletion of six nucleotides in the S gene, which results in loss of two amino acids at positions 69 and 70 (Δ69-70) and has been previously described by another group to cause S gene dropout in commercial assays (Bal, A. et al. medRxiv, 2020).
We discovered that Δ69-70 is present in >99% of sequenced S gene dropouts, but less than 0.1% of sequenced S gene positives from the same labs. We term this S-gene target failure (SGTF).
Further confirmation was provided by molecular analysis: sequencing of diagnostic PCR amplicon products from S gene dropout samples showed that the S gene target contains the Δ69-70 deletion in the middle of the amplicon. We infer that because the S gene is successfully amplified that the S gene dropout must be due to a failure of the qPCR probe to bind as a result of the Δ69-70 deletion.
Some variants other than the VOC also have Δ69-70, but as of late November, the VOC represents nearly all observed sequences with that mutation (Figure 2, Table 2).
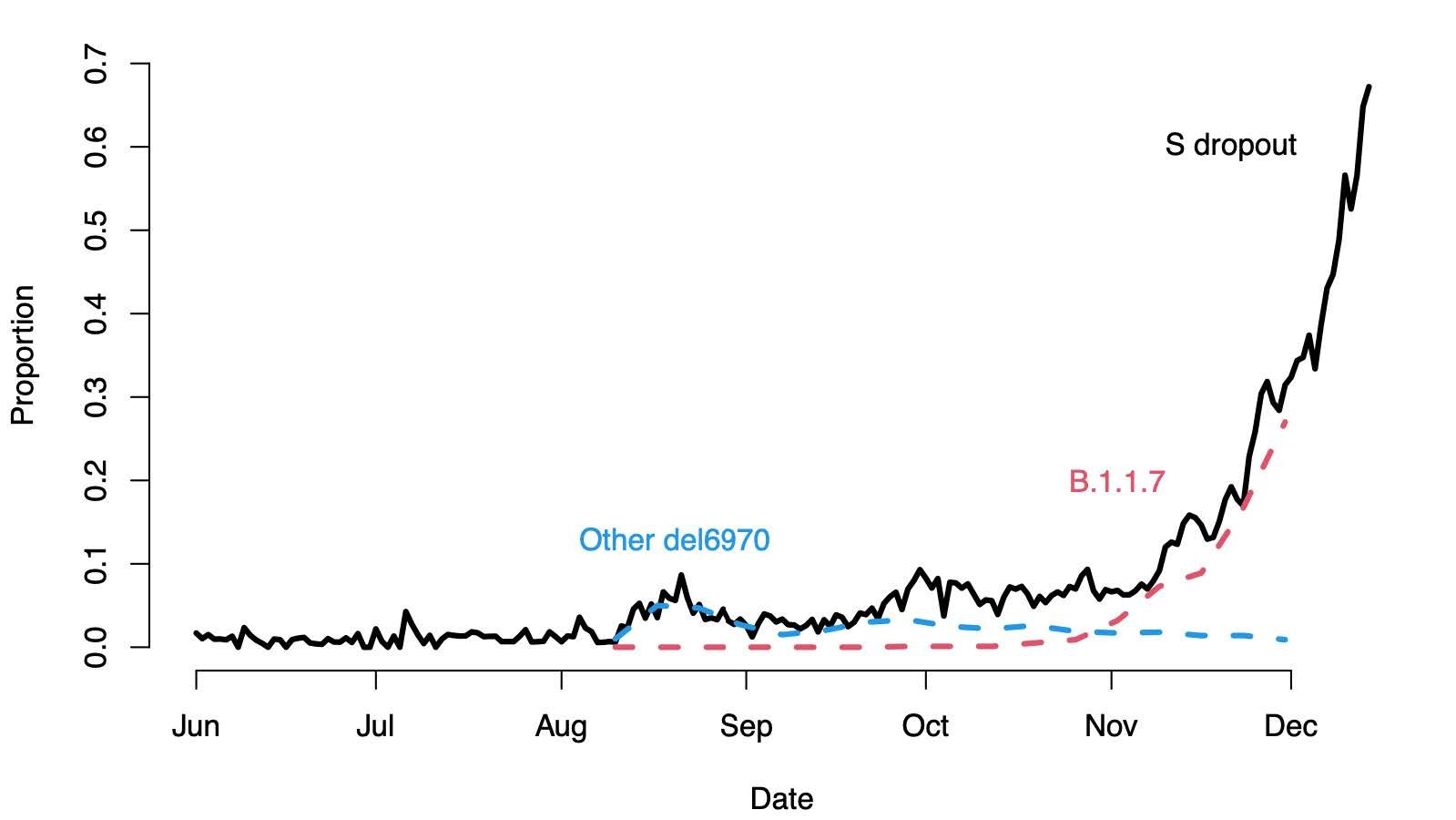
Figure 2. The solid black line shows the proportion of positive tests with S dropout at the Milton Keynes Lighthouse lab, the dashed red line shows the proportion of all Lighthouse sequences that are B.1.1.7, and the blue dashed line shows the proportion of sequences that are other variants with Δ69-70.
Table 2. Percent of all Pillar 2 Δ69-70 sequences by week that are the new variant, B.1.1.7.
| Week beginning | Percent new variant of all Δ69-70 |
| 2020-10-12 | 5% |
| 2020-10-19 | 15% |
| 2020-10-26 | 32% |
| 2020-11-02 | 54% |
| 2020-11-09 | 78% |
| 2020-11-26 | 86% |
| 2020-11-23 | 94% |
| 2020-11-30 | 96% |
Virological and phenotypic investigations
Virus is currently being isolated in laboratories at Imperial College, PHE Colindale and PHE Porton Down. No further biological data on antigenicity or in vitro replication or fitness is yet available.
PHE and partner laboratories are currently preparing early passage stocks of the variant. It is our aim to share these isolates with researchers as soon as feasible but it is likely that significant stocks of appropriately quality preparations will not be available until early in January. We plan to distribute to national laboratories via NIBSC and internationally through the European Virus Archive and others.
Summary
A novel variant has been identified which has spread rapidly within the UK. We have assessed this variant as having substantially increased transmissibility with high confidence. Further studies are underway to characterise the variant and updates will be provided.
Data sources
Data used in this investigation is routine data from the COG-UK dataset, PHE Second Generation Surveillance System and the PHE Rapid Investigation Team Kent investigation.
GISAID reference genome
Sequences from this VOC can be identified by searching for the B1.1.7 lineage on GISAID (gisaid.org). The canonical VOC genome is deposited with accession EPI_ISL_601443.
Supporting figures
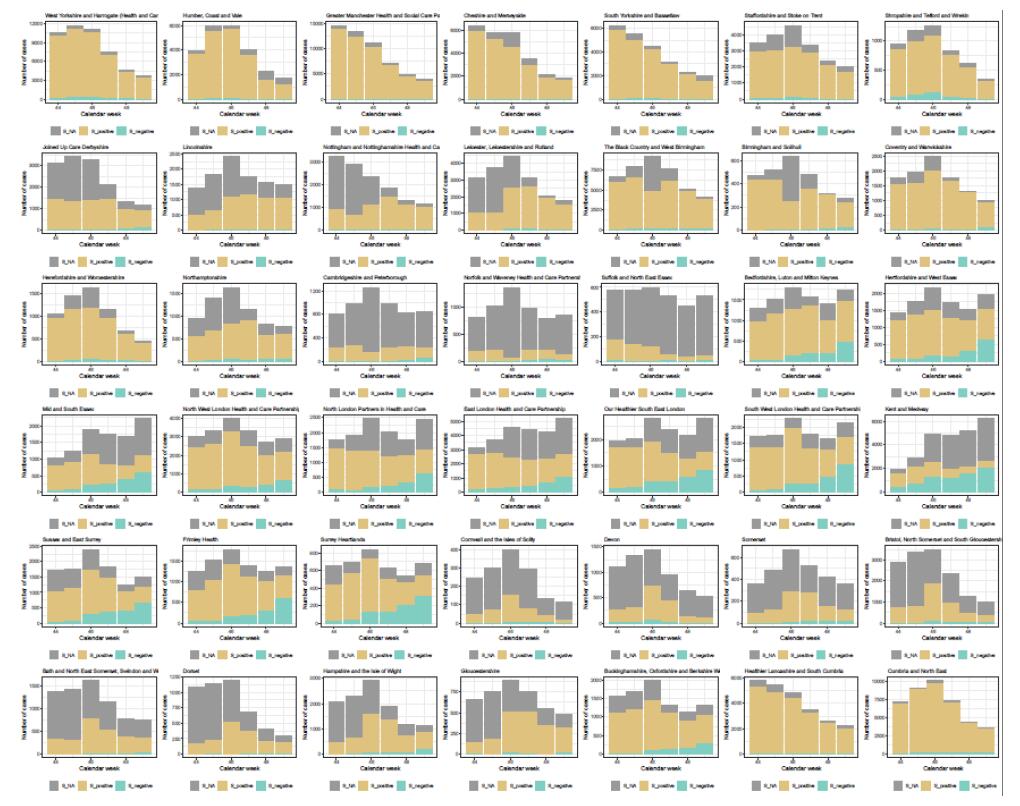
Figure S1. Pillar 2 case counts stratified by calendar week and NHS STP
area. Gold bars are S-gene positive, Cyan S-gene negative and gray bars are unspecified. The barplots show that for regions experiencing plateaus or growth the fraction of S-gene negative is increasing.
