




The Landscape of Antibody-drug Conjugate (ADC): Production, Mechanisms of Action (MOA), FDA approved-antibodies, and Functional assay
 Antibody-drug Conjugate (ADC) Products List Download
Antibody-drug Conjugate (ADC) Products List Download
|
Content index Antibody-drug Conjugate (ADC) Products Introduction Case study: Product data of Antibody-drug conjugate (ADC) GeneMedi's products list of Antibody-drug conjugate (ADC) What is antibody-drug conjugate (ADC)? —— Introduction Antibody-drug conjugate (ADC) in clinical application (Approved/BLA, phaseI/II/III) Main elements of Antibody-drug conjugate (ADC) 1. Antibodies and their targets of Antibody-drug conjugate (ADC) Review for Antibody-drug conjugate (ADC) production, quality control and functional assay |
Products Introduction
Antibody-drug conjugate (ADC) is a new generation of therapeutic drugs, consisting of antibodies, small molecule drugs (payloads), and linkers connecting antibodies and payloads. The navigators target specific tumor cells and the small molecules enter tumor cells to kill them. ADCs may serve as a novel therapeutic modality for many cancer patients. To meet the needs of ADCs development, GeneMedi provides more than 100+ ADC products.
Check the product information of Anti-Monomethyl auristatin E (MMAE) mouse monoclonal antibody with this link: https://www.genemedi.net/i/anti-monomethyl-auristatin-e-mmae-monoclonal-antibody
Case study: Product data of Antibody-drug conjugate (ADC)
| Antibody | Payload | QC |
|---|---|---|
| Ab-001 | VCMMAE | SDS-PAGE(reducing and non-reducing) |
| Human IgG1 Control | DAR | |
| Ab-002 | Cytotoxity assay |
First, we used a higher dose of reducing agent (TCEP) and small toxic molecule (vcmmae) to ensure the success of coupling. SDS-PAGE (reducing DTT & non reducing DTT) results showed that we successfully linked MMAE to the antibody. Here you are the conjugate manufacturing.
2.1 human IgG1-ADC: SDS-PAGE (reducing) and SDS-PAGE (non-reducing)
2.2 DAR detect
In this batch, we selected 5.4 equivalent TCEP and 10.8 equivalent VCMMAE groups for DAR detection. The DAR of our sample is almost the same as that of the standard sample, but the integrity of the antibody result is not as good as that of the standard sample.
2.3 cytotoxity assay
At next step, we did cytotoxity assay to observe the toxic effect of ADC on 293 cells. At the same time, considering the storage and transportation of ADC, we lyophilized ADC and compared the effect of non-lyophilized on ADC cytotoxicity.
Obviously, compared with the blank group, ADC had a significant inhibitory effect on the growth of 293 cells. Moreover, lyophilized can reduce the inhibition of ADC on cell growth.
The results show that when the equivalent of reducing agent TCEP is 0.4 and 1.5, the results do not have regularity, which may be caused by the limited opening of disulfide bond and the limited coupling of small molecules. However, when the equivalent of TCEP is 3, the results can match the previous PTM-1-ADC (5.4t5.4v & 5.4t10.8). We also deal 293-ko cell line with PTM-1-ADC, obviously, there is no regular relationship between cell death and ADC dose.

Figure. QC and functional verification of ADC(human IgG1-MMAE).
Figure A(SDS-PAGE, DTT) and figure B(SDS-PAGE, no DTT): Lane2 is human IgG1-naked; Lane3 is human IgG1 positive control. Lane4-7: Antibodies were treated with different equivalents of reductant (TCEP) and then added with different equivalents of conjugated small molecules (MMAE).
Figure C and figure D: LC-MS to detect the DAR(drug-to-antibody ratio) of ADC. Figure C is positive control and figure D is the ADC made by ourselves. Both are human IgG1-MMAE.
Figure E and figure F: ADC cytotoxicity assay.
GeneMedi's products list of Antibody-drug conjugate (ADC)
What is antibody-drug conjugate (ADC)? --Introduction
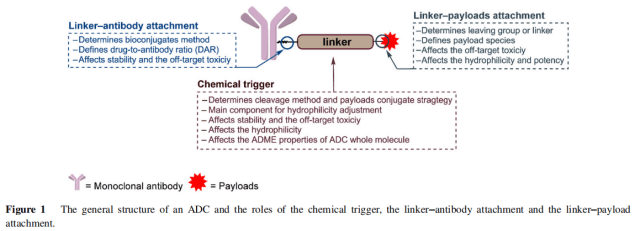
1. The structure of antibody-drug conjugate (ADC)
ADC consists of antibodies and payload, linker connects antibodies and small molecule drugs. After ADC drugs enter the blood, its antibody part will recognize and bind to the surface antigen of target cells. Theninternalizing ADC antigen complex into cells through endocytosis, the complex will be degraded by lysosomes and the payload will be released, so as to destroy DNA or microtubules, or exert the inhibitory effect of topoisomerase / RNA polymerase, resulting in cell death. It has the characteristics of precision and great lethality. It is a new generation of therapeutic drugs.1
2. Cellular Processing of antibody-drug conjugate (ADC)
ADC is composed of navigator targeting specific tumor cells and small molecules that can enter tumor cells to kill them, so how to maintain activity in blood to achieve good presentation effect is also a problem. Of course, accurate targeting can greatly reduce the sidekill effect. After ADC is swallowed into cells, lysosomal enzymes will hydrolyze the navigation of missiles (antibodies), thus releasing small cytotoxic molecules. Thus, it can block a series of activities in cells, including the transcription and translation of DNA and RNA. It can also block the transport of proteins by affecting tubulin. Finally, it can kill tumor cells. Therefore, here we need to pay attention to the consequences of Miss target and the loading of small molecules, which are potentially harmful to normal tissues and cells. The suspension of many clinical drugs in phase II and phase III is also greatly related to these two key points.
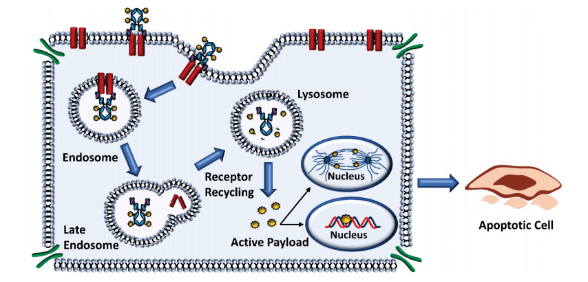 Figure 2. Cellular Processing of ADCs. Most ADCs undergo similar mechanisms to release the cytotoxic payload. In general, ADCs are designed for internalization and are processed via the endocytic pathway resulting in release of the payload and cytotoxic effect.2
Figure 2. Cellular Processing of ADCs. Most ADCs undergo similar mechanisms to release the cytotoxic payload. In general, ADCs are designed for internalization and are processed via the endocytic pathway resulting in release of the payload and cytotoxic effect.2
3. How antibody-drug conjugate (ADC) work? Mechanisms of action (MOA) for ADC
The drugs in ADC can recognize the membrane proteins of tumor cells, then locate the target cells. After endocytosis into cells, lysosomes hydrolyze and release small molecules in ADC. For example, VCMMAE is an anti-mitotic agent in antibody coupled drugs (ADC). It is crosslinked by monomethylalistatin e (MMAE) and dipeptide valine citrulline (VC). VCMMAE is a powerful anti-mitotic agent by blocking tubulin polymerization. VCMMAE effectively resist mitosis and kill cells by blocking tubulin polymerization. Microtubules play an important role in cell function, participate in migration, transportation and recombination, and have many dynamic roles, including the movement of motor proteins and the separation of chromosomes during cell division.
Antibody-drug conjugate (ADC) in clinical application (Approved/BLA, phaseI/II/III)
1. FDA approved Antibody-drug conjugate (ADC) for clinical use
Clinically, most of the ADC drugs approved by FDA are IgG1, and the targets are CD33, CD22, HER2 and so on. The most common payload is MMAE, also contains calicheamicin, DM1. ADC drugs are mainly used in the field of antitumor, which is one of the hot research directions in recent years. At present, 11 ADC drugs have been approved in the world, including Mylotarg (Pfizer), adcetris (Seattle genetics / Takeda), kadcyla (Roche), besponsa (Pfizer), lumoxiti (AstraZeneca), Polivy (Roche), padcev (Seattle genetics / anstelai / MSD), enhertu (AstraZeneca / first third party), trodelvy (immunomedicine), blenrep (GSK) Akalux (Rakuten Aspyrian)3. From the perspective of listed drug R & D enterprises, Pfizer, Seattle genetics, Roche and AstraZeneca have two models respectively, and the other three companies have one model respectively.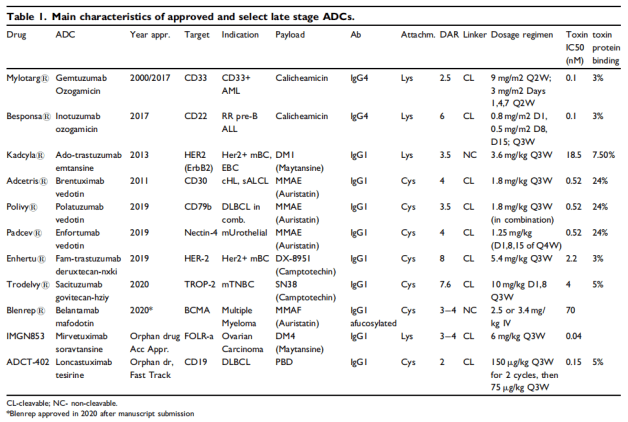
2. Antibody-drug conjugate (ADC) currently under clinical investigation
There are currently 82 novel ADCs in 150 active clinical trials registered with clinicaltrials.gov for cancer patients. Most of the ADCs are currently under investigation in phase 1 trials, while a small percentage has advanced to phase 3. Of the 150 ongoing trials, more than 80% are evaluating ADC safety and efficacy in solid tumors whereas less than 20% are trials for hematological malignancies. There are 43 disclosed targets organized here by the number of ADCs designed to recognize them. Most of these targets are under evaluation by a single ADC, while some are being investigated by several different ADCs. Of the 82 novel ADCs, followed by DNA-damaging molecules, topoisomerase I inhibitors, and finally unique payloads such as TLR agonists, a BCL2-xL inhibitor, and an RNA polymerase II inhibitor. Many payloads can’t be disclosed. Most ADCs under clinical investigation either utilize the conventional cysteine conjugation strategy or site-specific conjugation linker while few conjugate to surface lysines. 4
3. Novel Antibody-drug conjugate (ADC) in Clinical Trials
More than 80 ADCs are currently in active clinical trials, with a majority in phase I and I/II. Over 80% of the clinical trials are investigating ADC safety and efficacy in various solid tumors, while the remaining trials involve hematological malignancies. HER2 is currently one of the most attractive targets for ADC development, with three anti-HER2 ADCs currently in phase III trials. One such anti-HER2 ADC is RC48, produced by RemeGen, joining an IgG1 anti-HER2 antibody, hertuzumab, to approximately four MMAE molecules via a protease-cleavable valine-citrulline linker through cysteine conjugation.5
Main elements of Antibody-drug conjugate (ADC)
1. Antibodies and their targets of Antibody-drug conjugate (ADC)
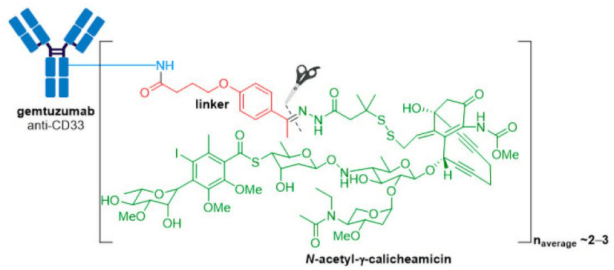
1.1 Mylotarg® (gemtuzumab ozogamicin) from Wyeth/Pfizer was the first ADC to reach the market. It is composed of a recombinant humanized anti-CD33 mAb (IgG4κ antibody hP67.6) covalently attached to a calicheamicin derived payload (N-acetyl-γ-calicheamicin 1,2-dimethyl hydrazine dichloride) via a pH-sensitive hydrazone linker.
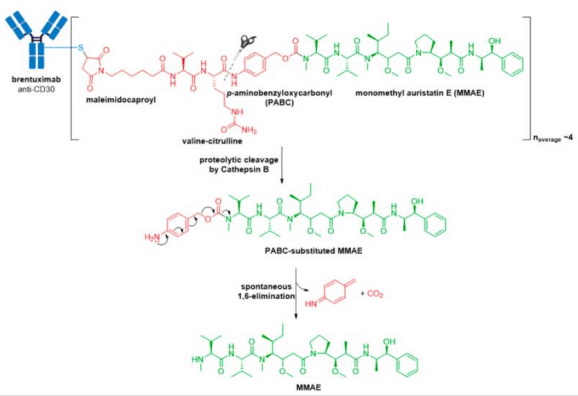
1.2 Adcetris® (brentuximab vedotin) from Seagen (formerly Seattle Genetics), containing a CD30-specific mAb conjugated to monomethyl auristatin E (MMAE), received FDA approval in 2011, making it the second ADC to enter the oncology market.
1.3 In 2013, Kadcyla® (ado-trastuzumab emtansine), developed and marketed by Genentech/Roche, revolutionized the field of ADCs by becoming the first ADC approved for the treatment of solid tumors. It is indicated as an adjuvant (after surgery) treatment for HER2+ early breast cancer in patients who previously received trastuzumab (Herceptin®) and a taxane, separately or in combination
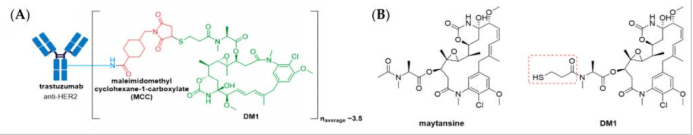
(A) Structure of Kadcyla® (ado-trastuzumab emtansine). The antibody is shown in blue, and chemical structures for linker and payload are in red and green, respectively. (B) The chemical structure for maytansine and DM1. The thiopropanoyl group of DM1, which allows for conjugation to a maleimidomethyl cyclohexane-1-carboxylate (MCC) group is shown in the red box.
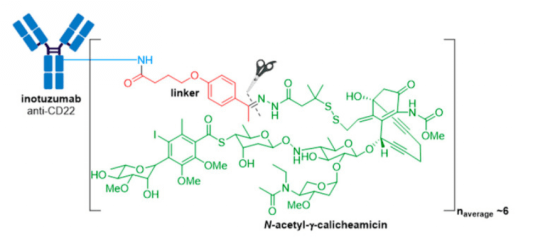
1.4 Besponsa® (inotuzmab ozogamicin (Pfizer/Wyeth)) obtained FDA approval in 2017 and is directed against CD22+ B-cell acute lymphoblastic leukemia (B-ALL). The first difference lies in the mAb and thus the antigen target and cancer indication. The recombinant humanized monoclonal IgG4 antibody (G544) employed in Besponsa® is selective for CD22 expressed on B cells in all patients with mature B-ALL, and >90% of patients with precursor B-ALL. n.
1.5 Polivy and Padcev Polivy® is an anti-CD79b ADC developed by Genentech/Roche using a proprietary technology developed by Seagen. It is indicated in combination with bendamustine and rituximab for treatment of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), an aggressive type of non-Hodgkin lymphoma, who have received at least two prior therapies. This indication was granted accelerated approval based on a complete response rate. Polivy® has an approximate DAR of 3.5 molecules of MMAE attached to each antibody.
Padcev®, produced and marketed by Astellas Pharma Inc. and Seagen is a Nectin4-directed ADC. It was first granted accelerated approval in 2019 for treatment of adults with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing therapy. In 2021, this indication was granted regular approval and Padcev® was granted accelerated approval for patients which are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy. Padcev® is comprised of a fully humanized anti-Nectin4 IgG1κ mAb (AGS-22C3) produced by mammalian (Chinese hamster ovary) cells, and has an approximate DAR of 3.8.
1.6 Enhertu® (fam-trastuzumab deruxtecan-nxki), developed by Daichi Sankyo/AstraZeneca, was granted accelerated FDA approval in December 2019 for treatment of adult patients with unresectable or metastatic HER2+ breast cancer who have received two or more prior anti-HER2 based regimens. The ADC is comprised of an anti-HER2 antibody, a protease cleavable tetrapeptide-based linker, and DXd as the drug payload.
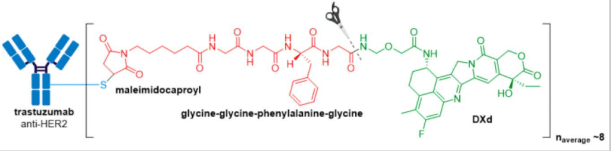
In April 2020, Trodelvy® received accelerated FDA approval for treatment of patients with locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received at least two prior therapies for metastatic disease. Trodelvy® consists of a fully humanized hRS7 IgG1κ antibody targeted against TROP2 (trophoblast antigen 2) conjugated to SN-38, the active metabolite of irinotecan via an acid-sensitive hydrolysable linker called CL2A.
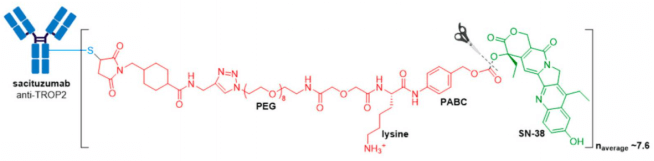
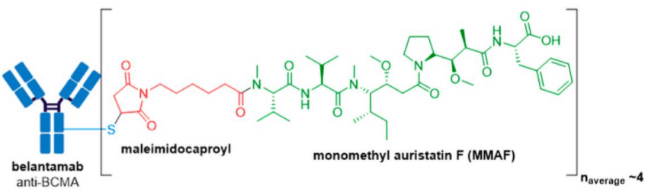
GlaxoSmithKline’s ADC, Blenrep® (belantamab mafodotin-blmf), is the first approved anti-BCMA (B-cell maturation antigen) therapy. It was granted accelerated FDA approval in August 2020 for treatment of adult patients with relapsed or refractory multiple myeloma who have received at least four prior therapies, including an anti-CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent. Blenrep® consists of an afucosylated humanized IgG1 mAb conjugated to the tubulin inhibitor, monomethyl auristatin F (MMAF) via a non-cleavable maleimidocaproyl linker. In addition to MMAF-induced apoptosis, secondary antitumor activity results from tumor cell lysis through ADCC and ADCP effector functions.
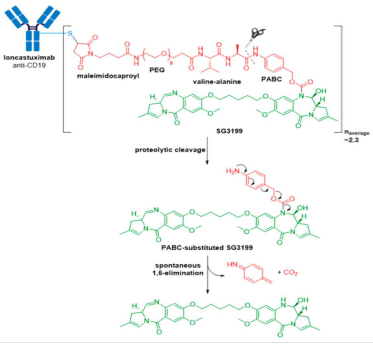
Zynlonta® (loncastuximab tesirine-lpyl) developed by ADC Therapeutics is a CD19-directed ADC indicated for treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL), not otherwise specified DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma. It was granted accelerated approval for medical use by the FDA in April 2021. Zynlonta® is composed of a humanized IgG1κ mAb conjugated to SG3199, a cytotoxic pyrrolobenzodiazepine (PBD) dimer alkylating agent, through a protease-cleavable valine-alanine linker.
In late September 2021, the FDA granted accelerated approval to Tivdak® (tisotumab vedotin-tftv), deeming it the most recently approved ADC on the market. Tivdak®, co-developed by Seagen and Genmab, is the first and only approved ADC indicated for treatment of adult patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy . Tivdak® is a Tissue Factor (TF) directed ADC comprised of a human anti-TF IgG1κ antibody conjugated to MMAE via the same protease-cleavable mc-vc-PABC linker construct employed in Adcetris®, Polivy®, and Padcev®. As for these previously discussed ADCs, Tivdak® carries an average of four MMAE molecules per mAb. Furthermore, in vitro studies have demonstrated that this ADC also mediates ADCP and ADCC effector functions, thus providing multimodal antitumor activity.
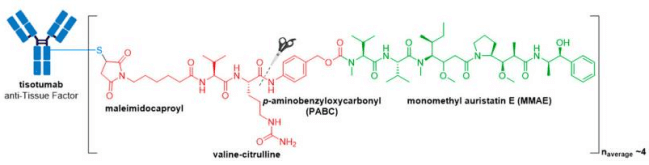
As ADCs have undergone clinical development, it has become clear that the rules applying to standard chemotherapy or antibody-based therapies on their own do not necessarily apply to ADCs. ADCs are modular in nature, with interchangeable components that can be altered in a strategic fashion to improve both their efficacy and toxicity profiles. 13
2. Linker (cleavable/non-cleavable, structure and mechanism) of Antibody-drug conjugate (ADC)
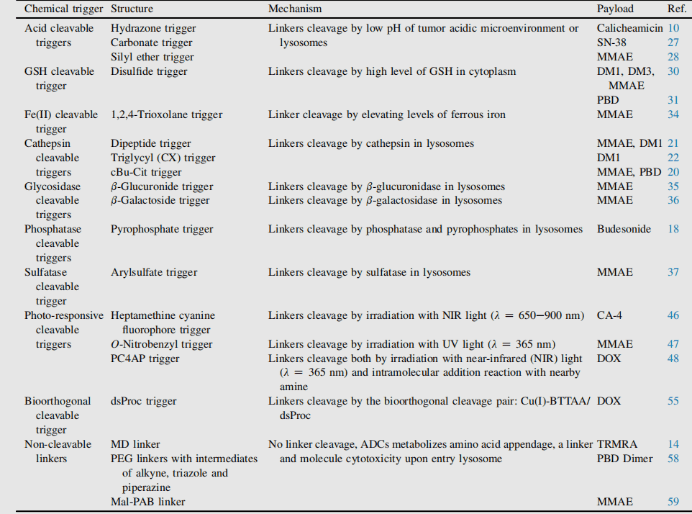
Table2. Chemical triggers
Conjugate linker is not only the molecular part forming covalent connection between antibody and small molecule payload, but also the key element with design properties in targeted drug therapy. The addition of linkers should not induce aggregation, and it is necessary to ensure acceptable PK characteristics, limit the premature release (stability) of payloads in plasma and effectively release active molecules at targeted action sites. In the process of connection, there are many conjugate companies. Connectors are divided into two types: non-cleavable linkers and cleavable linkers.6
ADC based on non-cleavable linker must be internalized, and the antibody part needs to be degraded by lysosomal protease to release active molecules. Many uncut linkers have been explored in the development of ADC. The most representative is n-succinimide-4 - (n-maleimide methyl) cyclohexane-1-carboxylate (SMCC), which is used by kadcyla.7
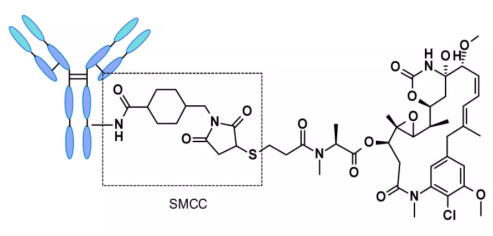
Catabolism of this structure leads to lys-smc-dm1 becoming the main tumor metabolite. In addition, drugs linked to this linker usually do not have a bystander killing/bystanerd effect, because the released catabolites have poor permeability. The current research mainly focuses on the cleavable linker.[11-12]
The use of cleavable linkers is equally feasible for the design of internalized and non internalized ADCs, because the release is triggered by the nature of the cleavage site (lysosome and / or tumor environment). Linkers can be divided into two categories: enzyme dependent and chemical (i.e. non enzyme) dependent (both have conjugate manufacturing).
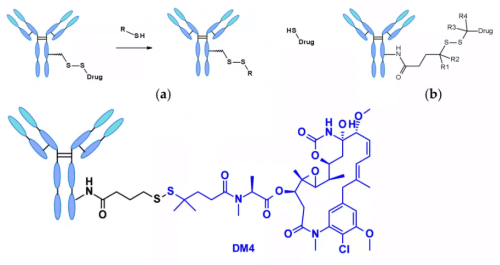
Chemically dependent linker : The linker containing disulfide bond is attacked by mercaptan to release the active load. Although the reduced form of human serum albumin (HSA) in plasma is the most abundant mercaptan, its reactivity to macromolecules is very poor.8The cytoplasm also contains high levels of glutathione (GSH), a tripeptide containing sulfhydryl, which is easy to react with s-nucleophilic protein. The difference of GSH concentration in blood (micro molar range) and cytoplasm (millimolar range) and oxidative stress caused by cancer cells contribute to the preferential release of drugs in cancer cells. Linkers containing disulfide bonds are mainly related to maytansinoid payloads. The reactivity of disulfide bonds can be adjusted by steric hindrance: α- Methyl substitution significantly affects the reduction rate and resistance to mercaptan disulfide bond exchange. For example, the linker of sar-3419 obtains the best antitumor activity of spdb-DM4 through dimethyl substitution.
Hydrazone linkers show pH dependent stability, stable at neutral pH, and hydrolyze in acidic medium (pH < 6 of endosomes and pH < 5 of lysosomes) to form corresponding ketones and hydrazine.
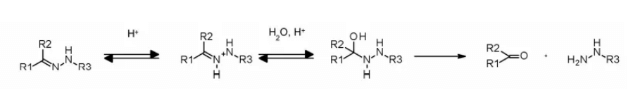
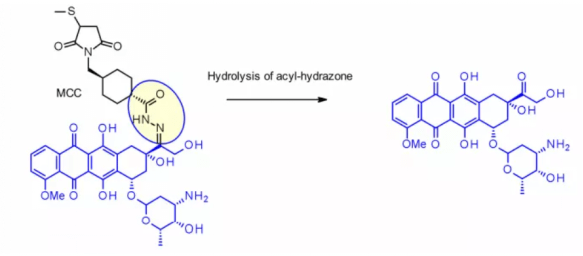
The method has been successfully applied to immu-110, which contains a cleavable acyl hydrazone linker, which is formed by the reaction of the hydrazide of 4-maleimide methylcyclohexane-1-carboxylate (MCC) with the ketone group present in adriamycin.
Hydrazone linkers are also often used for the payloads of the calimycin family. In this case, the release is triggered by a two-step activation process: the first step is that the acid sensitive hydrazone is hydrolyzed, and the second step is that the disulfide bond is reduced by GSH to cyclize the sulfhydryl intermediate. This linker has been used in the listed Mylotarg and besponsa, but their stability in plasma is not as stable as expected and not as attractive as other cleavable linkers.
Enzyme dependent linker: In order to limit the release of payload before internalization, so as to prevent or minimize the degradation of target cells, the protein components of lysosomes become a reasonable place to find enzymes that can degrade ADC and exist in high concentration.
Cathepsin-B
Cathepsin B is a cysteine protease, which exists in advanced endosomes and lysosomes of mammals and is overexpressed in many cancer cells. Initially, a cleavable dipeptide was used as the substrate of cathepsin B as adriamycin prodrug. This work established the dipeptide part of SAR: hydrophilic residues (citrulline or arginine) are required at P1, while lipophilic residues at P2 enhance plasma stability (phenylalanine, valine or alanine).
In addition, a self degradation spacer was introduced to promote the entry of the enzyme, thus limiting the steric hindrance of the payload: the spontaneous 1,6-elimination of p-aminobenzylcarbamate (PABA) in acidic medium, releasing carbon dioxide, p-azaquinone formamide and adriamycin. Finally, this discovery was transferred from the prodrug to the ADC field, demonstrating the antigen driven cellular activity of Val CIT and Phe Lys dipeptide linkers.9
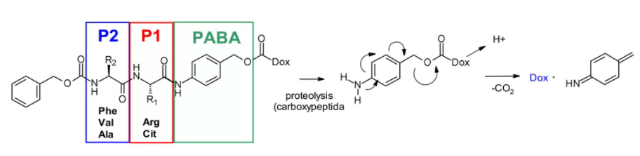
Val-Cit dipeptide is the most commonly used cleavable linker in ADCs. At present, there are up to 25 molecules in clinical stage, which may be due to its overall good plasma stability, release behavior and chemical tractability. Two approved ADC drugs (adcetris and polyvy) use the same linker MC VC PABC, which contains maleimide spacer, standard Val CIT dipeptide sequence as cathepsin substrate and PABC self degradation spacer.
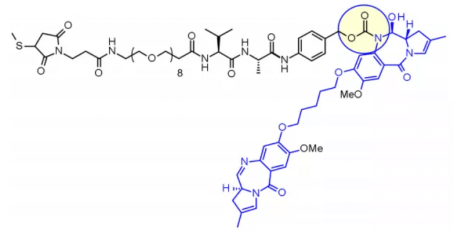
Val-Ala dipeptide is also widely used. Seven molecules are in the clinical stage. The fastest progress is loncastuximab tesirine, which includes a PEGylated spacer to balance the lipophilicity of the payload sg3199 belonging to the PBD dimer family.
Research shows that Val CIT is difficult to achieve high Dar due to precipitation and aggregation. In contrast, the Val ala linker allows Dar up to 7.4 with limited aggregation (< 10%). Compared with Val CIT, Val ala has lower hydrophobicity, which explains why this linker is excellent in lipophilic payload (such as PBD dimer). Val ala linkers of seven clinical candidate ADCs are connected to PBD.
Some studies have compared the structure of Val CIT and val ala dipeptides with the payload connection of MMAE. In the case of non internalized antibodies, Val CIT and val ala linkers bound to engineered cysteine showed similar
characteristics and better performance than Val Lys and val Arg analogues. In the case of anti HER2 ADC with random cysteine binding, Val ala showed less aggregation in the high Dar structure than Val CIT. On the other hand, the two linkers showed similar buffer stability, cathepsin B release efficiency, cell activity and histopathological characteristics.
Tetrapeptide Gly-Gly-Phe-Gly shows all the characteristics of stable and effective cleavable linkers, which are used by the listed ADC drug enhertu. The first trimester of enhertu is a plasma stable ADC with a dar of 7.7. Protease degradation occurs in lysosomes and dx-8951f is released. It is an effective topoisomerase I inhibitor derived from exatecan. Since the linker does not contain solubilizers, reaching such a high Dar is very considerable because it contradicts the widely established principle that high Dar conjugates may have poor pharmacokinetic characteristics. The self degradation spacer used here is simple and compact hemiamination, rather than the PABC used by Val-CIT linker.
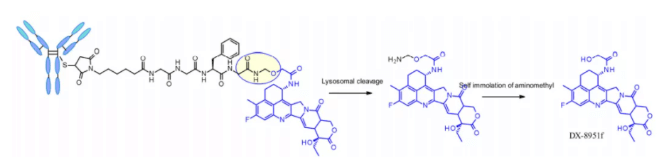
Phosphatase and pyrophosphatase
Like cathepsin, pyrophosphatase and phosphatase are hydrolases selectively expressed in lysosomes. In 2016, Merck researchers designed a linker containing phosphate and pyrophosphate to be paired with cathepsin b-sensitive Val CIT PABA to transfer glucocorticoids: phosphate / pyrophosphate partially binds between the self degrading spacer PABA and the payload. After internalization, the payload can be released in the order of cathepsin B, self degradation spacer and phosphatase (n = 1). For pyrophosphate (n = 2), another step involving pyrophosphatase may be required.
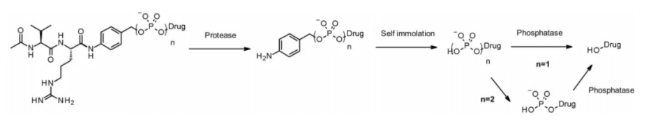
This hydrophilic and permanently charged group has the advantage of solubility. It can not only biocouple with lipophilic glucocorticoid derivatives, but also promote the purification of ADC. The residual linker in ADC is less than 0.10%. ADC containing phosphoric acid and pyrophosphoric acid has activity in vitro.
β- Glucuronidase
β- Glucuronidases are glycosidases that catalyze β- Hydrolysis of glucuronic acid residues, which are highly expressed in lysosomes and tumor stroma. Seattle genetics researchers published a groundbreaking work in 2006. Anti-CD70 ADC uses a linker containing glucuronic acid, which is attached to the self degrading spacer. This linker exhibits low levels of aggregation, high plasma stability, and strong in vivo efficacy.10
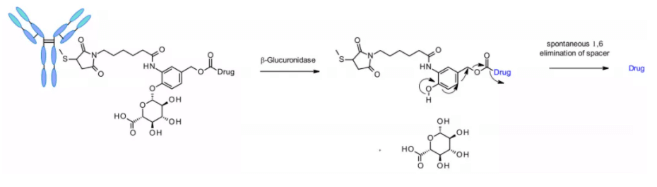
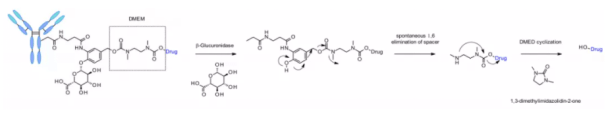
The linker is also applied to other amine containing payloads such as camptothecin analogues, sn38, ducamycin and matrine through an additional dimethylethylenediamine (DMED) self degradation spacer. Release sequence from hydrolysis β- From glucuronic acid to self degradation spacer, another cyclization reaction of DMED occurs spontaneously, forming 1,3-dimethylimidazoline-2-one, and finally releasing hydroxyl containing drugs. Due to the hydrophilicity of linker, compared with cathepsin sensitive linker, this technology makes the preparation of ADC DAR = 8 easier.
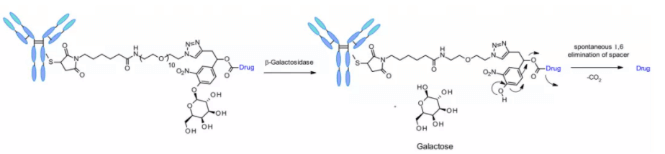
β- Galactosidase
Recently, a method of using β- Galactosidase cleaves the ADC of the linker, which contains the PEG10 spacer. The spacer was substituted by nitro to improve the self degradation rate. analogy β- Glucuronidase linker, whose dissociation mechanism involves hydrolysis β- Galactosidase moiety, which imparts hydrophilicity to chemical precursors. Another advantage is β- Galactosidase exists only in lysosomes, and β- Glucuronidase is expressed in lysosomes and in the microenvironment of solid tumors. Studies have shown that in the context of anti-HER2 ADCs releasing MMAE, it contains β- The ADC of galactosidase linker is more effective than t-dm1 in vitro and in vivo.
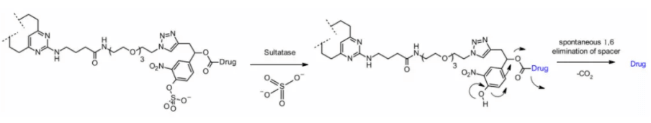
Sulfatase
Recently, there have been connectors cleaved by sulfatase, which is overexpressed in several cancer types and shows potential selectivity. The study involved anti-HER2 antibodies loaded with MMAE. Compared with the classical cleavable Val-Cit and Val-Ala linkers, the sulfatase linker showed similar efficacy on HER2+ cell lines.
3. Toxins/Payloads (Classification and function) of Antibody-drug conjugate (ADC)
There are so many payloads, like MMAE, Calicheamicin, MMAF, DM1, SN-38 and Dxd.
3.1. Microtubule destroying drug
a. Calendula
Auristatins is an important payload used in ADC. The most famous family member MMAE exists in two listed drugs, adcetris and Polivy. At present, more than 10 kinds of ADCs with calendula (such as MMAE) or methylcalendula f (MMAF) as payload are undergoing clinical trials.
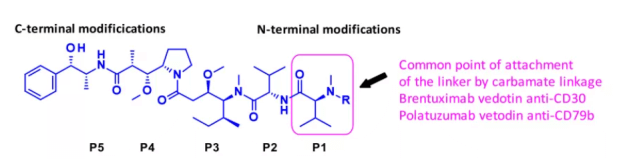
The figure above describes auristatine and its common connection sites. The structure-activity relationship (SAR) of Calendula has been widely studied, mainly focusing on the terminal subunit: P1 (N-terminal) and P5 (C-terminal). The most common method is to introduce carbamate function on P1.
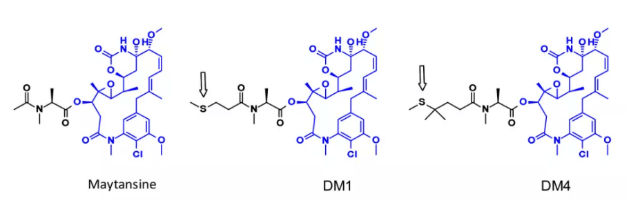
b. Meidengsu derivatives (DM2, DM4)
Metanthin is a very effective inhibitor of microtubule assembly, which can induce the cessation of cell mitosis. However, this structure is difficult to conjugate because it has no reactive functional groups. In order to overcome this problem, a series of very effective derivatives containing SME groups have been created. The first examples of such molecules are DM1 and DM4, which carry methylthiopropionyl rather than natural N-acetyl groups.
c. Microtubule lysin
Tubulysins is an effective inhibitor of microtubule polymerization, which can lead to the rapid disintegration of the cytoskeleton of dividing cells and apoptosis. They are a naturally occurring tetrapeptide family containing Mep, Ile, Tuv and Tut, R3 = OH or Tup, R3 = H.
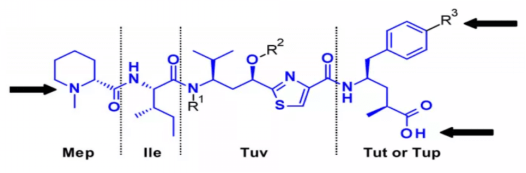
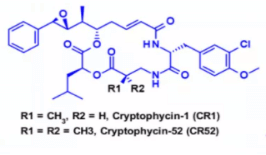
d. Cryptomyxin
Cryptomyxin (CR) is a family of six membered macrocyclic dipeptides with antitumor activity. The results of existing clinical trials show that the toxicity level is unacceptable at the dose required to achieve the therapeutic effect.
e. Anti-mitotic Eg5 inhibitor
Spindle kinesin (KSP, also known as Eg5 or kif11) is an ATP dependent motor protein involved in the separation of cell cycle centrosomes. Therefore, blocking this important event in mitosis with KSP inhibitors (kspis) can produce antitumor efficacy.
3.2 DNA damage drugs
a. Pyrrole benzodiazepines and indole chlorobenzodiazepines
Pyrrolo [2,1-c] [1,4] benzodiazepine (PBD) is a natural product with antitumor activity. Their mode of action is selective alkylation in small grooves of DNA, in which the N2 of guanine forms a covalent bond with the electrophilic N10 / C11 imine on PBD.
b. Ducamycin
Ducamycin is a powerful cytotoxic substance. It binds to the small groove of DNA through its highly active cyclopropane ring and alkylates adenine at N3 position. The non cyclized, halomethyl form of ducamycin significantly reduced its cytotoxic activity. Because the phenol group in the molecule can be used as the racemic activator to form electrophilic cyclopropane, the ligation strategy in the development of ducamycin ADC focuses on the linker ligation of phenol functional groups.
c. Camptothecin
Camptothecin (CPT) and its derivatives are classic examples of topoisomerase I inhibitors. They stabilize DNA single strand breaks induced by topoisomerase, and DNA double strand breaks occur when the ternary dna-top1-inhibitor complex encounters the replication fork. Natural camptothecin is a five ring structure. Its very low solubility prevents its wide application as a cancer therapeutic drug. Irinotecan, its water-soluble prodrug, has been licensed for the marketing of metastatic colorectal cancer. SN-38 is an active metabolite of irinoteptan. It is produced in vivo by the action of human liver carboxylesterase, which can be inactivated by opening the lactone ring.
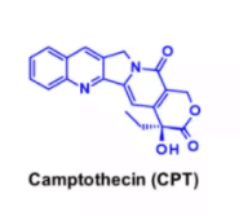
d. Kajimycin
Kajimycin is a widely studied class of enediyne antibiotics. Its structure and mechanism of action are particularly interesting and complex, making it a class of antibiotics in the field of ADC payload. The strategy of connecting calicheamicin in ADC takes ADC Mylotarg in the market as an example, and besponsa.
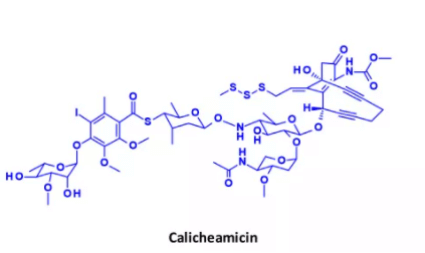
3.3 Innovative drugs
a. Apoptosis inducer (BCL XL inhibitor)
Overexpression of anti apoptotic Bcl-2 family members (including BCL XL) is one of the mechanisms for cancer cells to obtain apoptosis resistance. Drugs that block the BH3 binding domain on BCL XL can trigger cancer cell apoptosis.
b. Telanstadine and its analogues
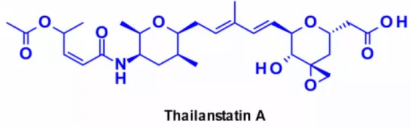
Targeted spliceosome is a large ribonucleoprotein complex involved in mRNA processing, which provides a promising treatment option for targeted cancer therapy. Several natural products can inhibit RNA splicing by binding to different splice subunits. The most representative is thailanstatin A, which can bind to the SF3B subunit of spliceosome to prevent RNA splicing.
c. Amanita toxin
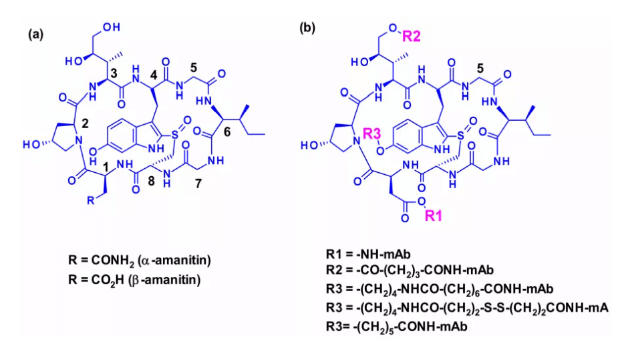
In the field of ADC technology, the use of transcription inhibitors similar to Amatoxins is a relatively new method. Nine naturally occurring amatoxin derivatives have the same skeleton structure. A macrocycle composed of eight L-configuration amino acids is partially connected between tryptophan and cysteine residues by sulfoxide. The three side chains of Amatoxins are hydroxylated, and the OH group has good water solubility and binds to the target molecule. Two peptides, α- Amanita glycoprotein and β- Amanita toxin accounts for 90% of all toxins.
d. Nicotinamide phosphoribosyltransferase
Nicotinamide phosphoribosyltransferase (Nampt) is an enzyme responsible for converting nicotinamide to nicotinamide mononucleotide. Its inhibitor has shown effectiveness in various preclinical and clinical studies, but its clinical application is limited by targeted toxicity and dose limiting toxicity, such as thrombocytopenia and gastrointestinal adverse reactions.
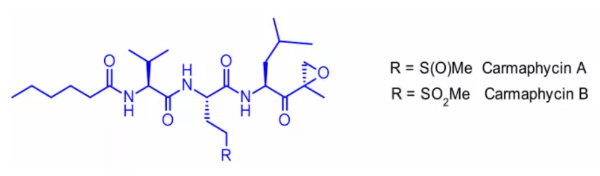
e. Camamycin
Two new protease inhibitors, camamycin A and camamycin B, were isolated from Curacao bacteria.
4. Coupling sites of Antibody-drug conjugate (ADC)
Biological coupling technology Chemical based specific in situ antibody modification
The natural structure of monoclonal antibodies provides a variety of possibilities for biological coupling. Chemical and specific natural (non engineering) antibody coupling has some advantages. It can avoid the complexity of antibody specific site mutation and the possible challenges in the amplification and optimization of cell culture.
Coupling sites according to the antibody sequence, the connection sites between disulfide bonds of endogenous amino acids such as lysine, histidine, tyrosine and cysteine are very attractive. All FDA approved ADCs use these endogenous amino acids for coupling until 2021. However, antibody scaffolds also contain glycans, which is caused by post-translational modification of Fc region during monoclonal antibody production. Some studies have reported new strategies for sugar engineering, which seems to be an interesting alternative to biological coupling.
Endogenous coupling of amino acids
One of the most common coupling methods is to use the lysine residue of the antibody, the amino acid nucleophilic NH2 group, to react with the electrophilic N-hydroxysuccinimide (NHS) Group on the lik payload. Although the reaction is simple, the high abundance of available lysine residues leads to the formation of uneven mixtures of many ADCs under random distribution. DAR is controlled by antibody-drug conjugates stoichiometry, which is widely used, including approved ADCs such as Besponsa, Mylotarg, and Kadcyla.
Disulfide re bridging strategy
IgG antibodies contain four disulfide bonds between chains, two connecting light chains and heavy chains, and two are located in the hinge region connecting two heavy chains. They maintain the integrity of monoclonal antibodies. Another classic biological coupling pathway explores the role of these cysteines as payload attachment points. The reduction of four disulfide bonds usually produces eight sulfhydryl groups, which can react with the linker of maleimide to produce ADC with DAR=8.
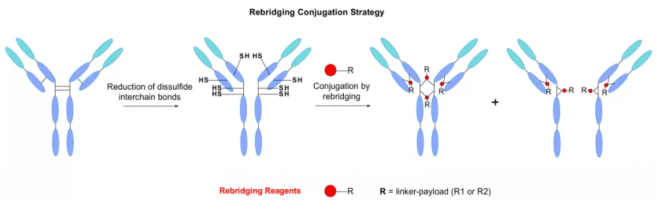
Dorona and colleagues reported an example of an ADC with a chimeric anti-CD30 monoclonal antibody coupled to MMAE, DAR= 8. Compared with the classical lysine coupling, this payload loading method is better controlled. However, it is reported that the plasma clearance rate will be higher and the risk of plasma aggregation will be reduced.
In 2015, chudasama et al. Introduced a new type of re bridging reagent, dibromopyridazinediones. They proved that it can be effectively inserted into the disulfide bond, and the resulting structure shows excellent hydrolytic stability even at high temperature. However, with the increase of temperature on the reduction step, heterogeneity is also observed, and this structure also allows the selective introduction of different functional groups.
Divinylpyrimidine is another effective re-bridging reagent, which can produce stable ADC with Dar = 4. Spring et al. Studied the effect of vinyl heteroaryl scaffold on cysteine re bridging. They believe that replacing pyridine with pyrimidine can make heteroaryl ring a better electron acceptor, so as to improve the crosslinking efficiency. Their work extends to divinyltriazine, which shows higher efficiency at high temperature.
In order to avoid the disadvantage of in vivo instability associated with classical maleimide coupling, Barbas et al studied methylsulfonylphenyloxadiazole, which has a specific reaction to cysteine. They are more stable than cysteine maleimide conjugates in plasma. Inspired by this, Zeglis designed dipods reagent, which contains two oxadiazole methyl sulfone parts connected by phenyl. Dipods forms covalent bonds with two sulfate radicals in the way of re bridging. Compared with maleimide coupling, coupling in this way has superior stability in vitro and performance in vivo.
Glycan coupling
Because IgG is a glycoprotein, it contains an N-glycan at n297 of CH2 domain of each heavy chain of Fc fragment. This glycosylation can be used as the attachment point of connecting payload. The long-distance localization between polysaccharide and Fab region reduces the risk of damaging the antigen binding ability of antibody after coupling. In addition, compared with the peptide chain of antibody, their chemical composition is different, allowing site-specific modification to make them suitable coupling sites.
Glycan biocoupling can be distinguished according to the technology used to target carbohydrates: including glycan metabolic engineering, glycan oxidation after glycotransferase treatment, endoglycosidase and transferase treated ketone or azide labeling.
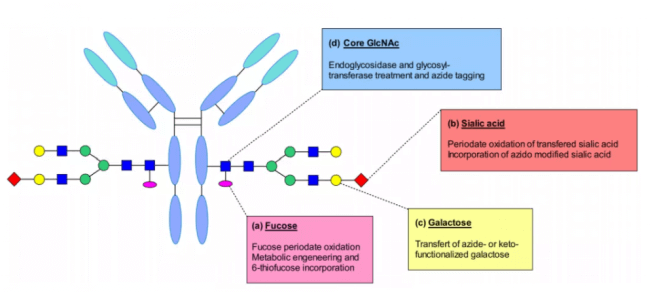
Neri et.al reported site-specific modification of fucose at the N-glycosylation site of IgG antibody. This sugar contains a cis diol moiety suitable for selective oxidation. They use sodium metaiodate to oxidize fucose residues to form an aldehyde group that can react with hydrazine containing linkers, so that the antibody is connected to the drug through hydrazone bonds.
Senter and colleagues added sulfur based analogues to the cell culture medium to bring 6-thiofucose into antibody modification through metabolism. They believe that substitution is accomplished by hijacking the fucosylation pathway, which introduces chemical sites to achieve site-specific binding. Compared with classical cysteine conjugates, this method significantly reduces the level of heterogeneity and produces conjugates with more predictable pharmacokinetic and pharmacodynamic properties.
Sialic acid is rarely contained in recombinant IgG. However, it has been proved that glycine can be enzymatically modified by galactosyl and sialyltransferase. Galactose was added by enzymatic reaction to obtain G2 glycan, and then terminal sialic acid was added. This modification generates aldehyde groups through periodate oxidation, which can couple linker payloads with hydroxylamine groups. The conjugate has high targeting selectivity and good antitumor activity in vivo. Periodate can also oxidize sensitive amino acids such as methionine and affect the binding with FcRn.
Site specific biological coupling of engineered antibodies
Advances in bioorthogonal chemistry and protein engineering contribute to the generation of more uniform ADCs. Although there are many attachment methods available on natural monoclonal antibodies, site-specific biological coupling on engineered antibodies can more effectively control Dar and avoid changing the affinity for antigen binding. In this way, natural or unnatural amino acids are added at some positions to obtain homogeneous products with excellent pharmacokinetic and pharmacodynamic characteristics.
Enzymatic method
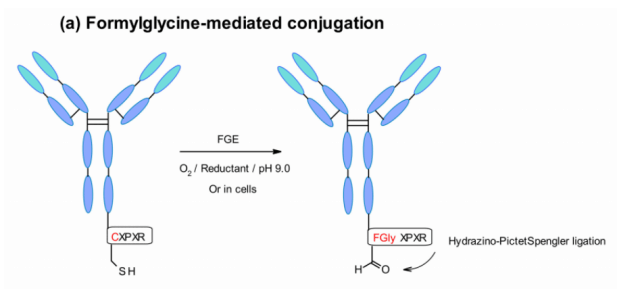
The attachment of payloads can be achieved in a very selective manner by inserting specific amino acid tags into the antibody sequence. These tags are recognized by specific enzymes, such as formylglycine generating enzyme (FGE), microbial transglutaminase (MTG), transpeptidase or tyrosinase, so that site-specific coupling can be performed.
Aaron et al. Explored a new site-specific coupling of aldehyde labeled proteins. The technique utilizes a gene encoded pentapeptide sequence (Cys-X-Pro-X-Arg), in which cysteine residues are recognized by FGE and co translated and oxidized to formylglycine during protein expression in cells. In this way, the engineered antibody is selectively coupled with aldehyde specific linker by hips (hydrazino Pictet – Spengler) chemical method.
Microbial transglutaminase (MTGase) strategies are also often developed for location-specific coupling. MTGase catalyzes the formation of peptide bonds between the glutamine side chain at the 295 position of the glycosylated antibody and the primary amine of the substrate. Compared with other enzyme strategies, MTG is a flexible technology and does not require peptide donors to achieve coupling. As long as the acyl receptor contains a primary amine, there is no structural restriction.
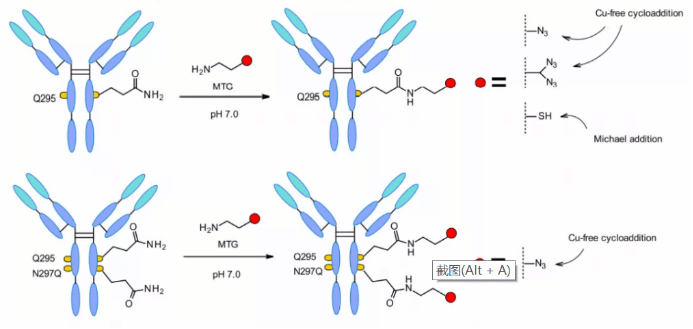
Glutamine residues naturally exist in the Fc region of each heavy chain of the McAb. After deglycosylation at position 295, glutamine residues are coupled through MTGase mediated reaction to produce a uniform ADC with Dar = 2. In order to improve the efficiency, the linker with branched chain can be coupled to double the Dar, and the mutation of asparagine at position 297 to glutamine can also increase the DAR.
NBE therapeutics developed S-based Staphylococcus aureus transpeptidase A-mediated coupling. Their strategy uses transpeptidase a (srtA) to cut the amide bond between threonine and glycine residues in the motif of lpxtg (X = any amino acid) pentapeptide. It then catalyzes the coupling of glycine related payloads with the newly formed C-terminal to form peptide bonds at physiological temperature and pH.
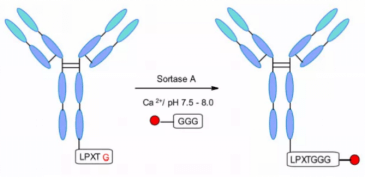
The method was applied to different antibodies, such as anti-CD30 and anti-HER2, and maytanine and MMAE were coupled with 5-glycine labeled linkers. Both ADCs showed similar in vitro cytotoxicity to classical coupling. Trastuzumab maytanine produced by enzymatic method completely matched kadcyla in vivo test.In another example, the ADC of efficient anthracycline toxin derivative pnu-159682 was generated by transpeptidase method. Interestingly, through this technology, the coupling efficiency is even higher than that of adcetris and kadcyla analogues. In addition, the prepared pnu-159682 ADC has high stability in vitro and in vivo, and shows more efficacy than the ADC containing tubulin targeting payload.
Biological coupling with engineered unnatural amino acids
In addition to thiomonoclonal antibody technology, the addition of non-standard amino acids (NCAA) provides another possibility for site-specific coupling. The technology uses amino acids with unique chemical structure, which can introduce linker payload complexes in a chemically selective manner. This technique requires the recombination of antibody sequences, using tRNA and aminoacyl tRNA synthetase (AARS) orthogonal to all endogenous tRNAs and synthetases in the host cell to bring NCAA into the protein in response to unassigned codons. Generally, NCAA is added to the medium during fermentation. The selection of unnatural amino acids is important because they may stimulate immunogenicity. The commonly used NCAA is an analog of natural amino acids with unique groups, such as ketones, azides, cyclopropenes or dienes.
Review for Antibody-drug conjugate (ADC) production, quality control and functional assay
1. SDS-PAGE
We need run SDS-PAGE(reducing and non-reducing DTT) to see the integrity of antibody and Preliminary observation of the connection of small molecules. In general, we can see that the conjugated antibody will shift upward compared with the naked antibody
2. DAR (methods and standard)
ADC drugs are essentially a mixture, which is composed of mAbs connecting different numbers of small molecule drugs. Dar represents the average number of small molecule drugs connected to each mAb. Dar directly affects the efficacy and safety of ADC drugs. In the drug development stage, the variation range of DAR value should be minimized. The coupling sites of ADC drugs are divided into amino groups on lysine residues and sulfhydryl groups on cysteine residues. The DAR coupled by lysine is often small, but there are many potential coupling sites. The coupling reaction is random and the product heterogeneity is large; There are 4 pairs of inter chain disulfide bonds used in the development of ADC drugs. The antibody converts the inter chain disulfide bond into free cysteine residues through partial reduction. The sulfhydryl group in the cysteine residues reacts with the maleimide group in the linker to form ADC. Currently, most ADCs in use or under development rely on interchain disulfide cysteines for conjugation, in which the 4 (IgG1 and IgG4) or 6 (IgG2) interchain disulfide bonds are reduced by an excess reducing agent, namely tris(2-carboxyethyl)phosphine or dithiothreitol.153 This spares disruption of intrachain disulfide bonds while freeing sulfhydryl groups from cysteine residues participating in interchain disulfide bonds (Figure 3e). The resulting product is a mixture of ADCs containing 0–8 drugs per parent IgG1 or IgG4 and 0– 12 per IgG2, with predominantly even numbered DAR (0, 2, 4, 6, 8, 10, 12) species within the ADC mixture.
2.1 Ultraviolet / visible spectroscopy (UV / VIS)
UV / Vis spectroscopy is the simplest and stable method to detect Dar value. This method requires antibodies and small molecule drugs to have different maximum absorption wavelengths, and calculate their concentrations respectively to obtain the DAR value of ADC, which is suitable for a variety of ADCs.
2.2 Chromatography
Chromatography includes hydrophobic interaction chromatography (HIC) and reverse phase high performance liquid chromatography (RP-HPLC), which are suitable for the determination of cysteine coupled ADC. Hydrophobic interaction chromatography can separate the components with different Dar values according to the difference of hydrophobicity, and maintain the structural integrity of ADC molecules; Reversed phase high performance liquid chromatography needs to reduce the antibody to obtain light and heavy chains before analysis. It can be used to supplement and verify the results of hydrophobic interaction chromatography, and is suitable for mass spectrometry.
2.3 Mass spectrometry
Mass spectrometry is suitable for the determination of DAR value of lysine coupled ADC, including liquid chromatography tandem mass spectrometry and MALDI-TOF-MS. Lysine coupled ADC has strong heterogeneity, which increases the difficulty of mass spectrum analysis. Usually, additional pretreatment of ADC is required before determination, such as deglycosylation and removal of C-terminal lysine heterogeneity.
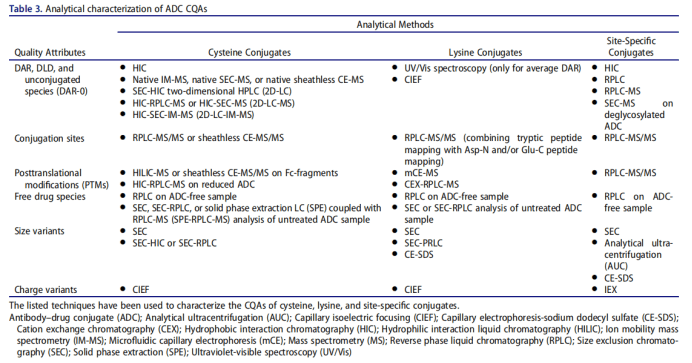
3. Cytotoxity assay of ADC
After the DAR of antibody is determined, the cytotoxicity experiment of ADC needs to be carried out. The cells are treated with different doses of ADC and their EC50 is analyzed. In the cytotoxicity experiment in vitro, the cells in the control group knocked out the target gene, so the antibody lost the role of localization and could not kill tumor cells, so it did not have the regularity of dose. However, from the treatment results of cells that have never been knocked out, they generally have better killing and EC50.
Reference
1. Su Z , Xiao D , Xie F , et al. Antibodydrug conjugates: Recent advances in linker chemistry[J]. Acta Pharmaceutica Sinica B, 2021.
2. Dean AQ, Luo S, Twomey JD, Zhang B. Targeting cancer with antibody-drug conjugates: Promises and challenges. MAbs. 2021 Jan-Dec;13(1):1951427. doi: 10.1080/19420862.2021.1951427. Erratum in: MAbs. 2021 Jan-Dec;13(1):1966993. PMID: 34291723; PMCID: PMC8300931.
3. Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao NS, Tice DA, Soria JC. Antibody-Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin Cancer Res. 2019 Sep 15;25(18):5441-5448. doi: 10.1158/1078-0432.CCR-19-0272. Epub 2019 Apr 12. PMID: 30979742.
4. Abedi M , Cohan R A , Mahboudi F , et al. MALDI-MS: a Rapid and Reliable Method for Drug-to-Antibody Ratio Determination of Antibody-Drug Conjugates[J]. Iranian biomedical journal, 2019, 23(6).
5. Xuejing, Yao, Jing, et al. A novel humanized anti-HER2 antibody conjugated with MMAE exerts potent anti-tumor activity[J]. Breast Cancer Research&Treatment, 2015.
6. Su Z , Xiao D , Xie F , et al. Antibodydrug conjugates: Recent advances in linker chemistry[J]. Acta Pharmaceutica Sinica B, 2021.
7. Chen Y , Kim M T , Zheng L , et al. Structural Characterization of Cross-Linked Species in Trastuzumab Emtansine (Kadcyla)[J]. Bioconjugate Chemistry, 2016.
8. Seki H , Walsh S J , Bargh J D , et al. Rapid and robust cysteine bioconjugation with vinylheteroarenes. 2021.
9. Kostova V, Désos P, Starck JB, Kotschy A. The Chemistry Behind ADCs. Pharmaceuticals (Basel). 2021;14(5):442.
10. Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442.
11. Singh A P , Sharma S , Shah D K . Quantitative characterization of in vitro bystander effect of antibody-drug conjugates[J]. J Pharmacokinet Pharmacodyn, 2016, 43(6):567-582.
12. Aleksandr V. Yurkovetskiy;Natalya D. Bodyak;Mao Yin;A Novel Antibody–Drug Conjugate Platform Featuring High Drug Loading and a Controlled Bystander Effect
13. Mol Cancer Ther (2021) 20 (5): 885–895.
14. Tong JTW, Harris PWR, Brimble MA, Kavianinia I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules. 2021;26(19):5847. Published 2021 Sep 27. doi:10.3390/molecules26195847
