


CRISPR/Cas9-gRNA system-mediated gene editing/gene knockout study and gene editing therapy
 CRISPR/Cas9 protocol Download
CRISPR/Cas9 protocol Download
Gene editing, also called genome editing, is a group of technologies to change the sequence of DNA in the genome. Several approaches to genome editing have been developed, including Zinc Finger, TALEN, and CRISPR/Cas9. Compared with the other gene editing tools, CRISPR/Cas9 system is faster, cheaper, more accurate, and more efficient, showing unprecedented advantages in gene therapy.
CRISPR/Cas9 premade products
| Cat No. | Products Name | Type of Crispr | Viral vector | Promoter | Fluorescent/Resistance | Tag | Order |
| GMV-Crispr-AAV001 | AAV-CMV-saCas9 | saCas9 | AAV | CMV | Null | HA |  |
| GMV-Crispr-AdV001 | Ad-CMV-spCas9 | spCas9 | Adenovirus | CMV | zsgreen | FLAG | |
| GMV-Crispr-AdV002 | Adv-CMV-spCas9-zsgreen | spCas9 | Adenovirus | CMV | EGFP | FLAG | |
| GMV-Crispr-AdV003 | Adv-CMV-spCas9-EGFP | spCas9 | Adenovirus | CMV | Null | FLAG | |
| GMV-Crispr-AdV004 | Adv-CMV-spCas9-mcherry | spCas9 | Adenovirus | CMV | mcherry | FLAG | |
| GMV-Crispr-LV001 | Lv-CMV-spCas9-puromycin | spCas9 | lentivirus | CMV | puromycin | FLAG | |
| GMV-Crispr-LV002 | Lv-CMV-spCas9-zsgreen | spCas9 | lentivirus | CMV | zsgreen | FLAG | |
| GMV-Crispr-LV003 | Lv-CMV-spCas9-EGFP | spCas9 | lentivirus | CMV | EGFP | FLAG | |
| GMV-Crispr-LV004 | Lv-CMV-spCas9-mcherry | spCas9 | lentivirus | CMV | mcherry | FLAG | |
| GMV-Crispr-LV005 | Lv-CBH-spCas9-puromycin | spCas9 | lentivirus | CBH | puromycin | FLAG | |
| GMV-Crispr-LV006 | Lv-CBH-spCas9-zsgreen | spCas9 | lentivirus | CBH | zsgreen | FLAG | |
| GMV-Crispr-LV007 | Lv-CBH-spCas9-EGFP | spCas9 | lentivirus | CBH | EGFP | FLAG | |
| GMV-Crispr-LV008 | Lv-CBH-spCas9-mcherry | spCas9 | lentivirus | CBH | mcherry | FLAG | |
Custom made CRISPR/Cas9 service
CRISPR/Cas9 User Manual
| Crispr/cas9 mediated Gene knockout
|
AAV Production
CRISPR/Cas9 Knockout System-User Manual |
Adenovirus
CRISPR/Cas9 Knockout System-User Manual |
Lentivirus-CRISPR/Cas9 Knockout System-User Manual |
Content of CRISPR/Cas9-gRNA system
- 1. Introduction and principles of CRISPR gene editing system
- 2. CRISPR/Cas9 delivery system
- 3. Protocol of CRISPR /Cas9 mediated gene knockout in vitro and in vivo
- 4. gRNA design and validation
- 5. Gene editing therapy
- 6. Reference
Introduction and principles of CRISPR gene editing system
CRISPR (clustered regularly interspaced short palindromic repeats) is a family of DNA sequences derived
from viruses that have previously infected the prokaryotes and has the ability to recognize invasive
homologous DNA sequences from similar viruses, then directing Cas9 (CRISPR-associated 9) nuclease to
specifically cleave them during subsequent infections, thus playing a key role in the antiviral defense
system of prokaryotes [4]. Cas9 nuclease is an enzyme guided by CRISPR sequences to recognize and cleave
specific nucleic acids which are complementary to the CRISPR sequence [5]. CRISPR sequences together with
Cas9 enzymes make up the basis of a CRISPR/Cas9 technology which is a versatile genome-editing platform
within organisms.
By 2010, three CRISPR/Cas9 systems have been identified in bacteria: type I, II and III. Derived from
Streptococcus pyogenes, type II CRISPR system is relatively simple and has been eventually adapted for
genome editing in mammalian cells [1]. This simpler CRISPR/Cas9 system contains four components, i.e. Cas9
endonuclease, trans-activating CRISPR RNA (tracrRNA) and two small RNA molecules named CRISPR RNA (crRNA)
[2]. In this system, the mature crRNA base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA
structure, guiding Cas9 nuclease to mediate the recognition and cleavage of target nucleic acids [3-5]. At
sites complementary to the crRNA-guide sequence, the HNH nuclease domain of Cas9 cleaves the complementary
strand, whereas the RuvC-like domain of Cas9 cleaves the non-complementary strand. To target nucleic acids
with more precision and expand the application of CRISPR/Cas9 system, great efforts have been paid to
engineer the Cas9 enzyme or alter PAM specificities. To date, a wide range of Cas9 derivatives with novel
PAM specificities have been generated using the site-depletion assay [6], and the initial two-RNA structure,
ie. CRISPR RNAs (crRNAs) base-paired to trans-activating crRNA (tracrRNA), have been programmed into a
"single-guide RNA (sgRNA)" (dual-tracrRNA:crRNA) by fusing the 3′ end of crRNA to the 5′ end of tracrRNA
[7]. By manipulating the nucleotide sequence of the sgRNA, the artificial CRISPR/Cas9 system could be
programmed to target any DNA sequence for cleavage [7].
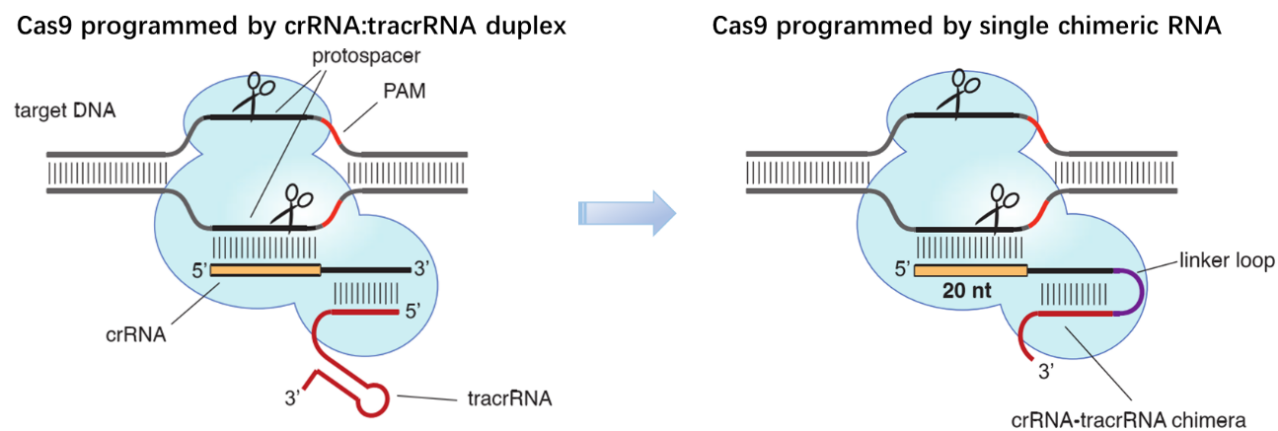
Figure 1. Cas9 can be programmed using a single engineered RNA molecule covering tracrRNA and crRNA
features. Left, Cas9 is directed by a dual RNA structure formed by activating tracrRNA and targeting crRNA
to cleave site-specifically–targeted dsDNA in type II CRISPR/Cas systems. Right, a chimeric RNA generated
through fusion of crRNA to tracrRNA [7].
1) CRISPR/Cas9 gene editing system
a. Cas9: I) NHEJ mediated gene knockout; II) HDR mediated gene knockin
Guided by small guide RNA (sgRNA), Cas9 nuclease cleaves double-strand DNA, the inducing site-specific
genomic double-strand breaks (DSBs). In mammalian cells, there are 2 manners with which DSBs are repaired:
non-homologous end joining (NHEJ) or homology directed repair (HDR). (I) Frequently, NHEJ happens, enabling
the introduction of small deletions or insertions between the ends of DSBs and resulting in frameshift
mutations, and thus mediates gene knockout [8,9]. (II) With the existence of repair template, such as
exogenous donor vectors, HDR takes place, introducing precise gene modifications, such as reporter
insertions, and thus mediate gene knockin [10,11]. Both NHEJ mediated gene knockout HDR mediated gene
knockin have been widely used in the field of gene therapy [12]. However, HDR (0.5%-20%) is much less
frequent than NHEJ (20%-60%) [13], and the former only occurs during S and G2 phase while the latter can
occur at any time of cell cycle [14-16]. To improve the efficiency of HDR, people have screened so many
small molecules, finding that small molecules inhibitor Scr7, targeting DNA ligase IV, one key enzyme in the
NHEJ pathway, significantly facilitates the HDR pathway (5%-58.3%) [17]. So far, NHEJ is widely used in the
mediation of gene knockout [8,18] and correction of pathogenic gene mutation [12,19,20], while HDR is
applied for knockin schemes, such as knockin cell line generation [21] or reporter/tag knockin [22].
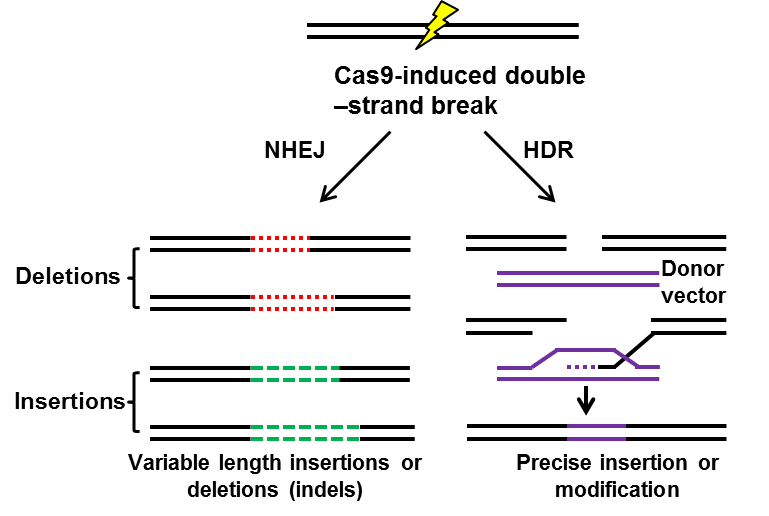
Figure 2. A schematic model of CRISPR system-induced non-homologous end joining (NHEJ) or homology
directed repair (HDR) [23,24].
b. Cas9 nickase: off-target and D10A
Although CRISPR/Cas9 system is much cheaper, more efficient and flexible, high efficiency of off-target
mutations or unwanted DNA cleavages have limited the application of this system. Mutation exploration
revealed that Cas9 nickase carrying the mutation of D10A (D10A Cas9) loses the nuclease activity in RuvC
domain, but does not affect the nuclease activity of HNH domain. In combination with paired gRNAs, D10A Cas9
cuts target DNA and generates two single-strand breaks with higher specificity and little off-target
mutations, which is confirmed by exome sequencing analysis [25,26]. By comparing gene-disrupting efficiency
of Cas9 paired nickases with that of nuclease, the T7E1 assay and deep sequencing revealed comparable
on-target efficiency, even sometimes higher on-target efficiency [27]. Moreover, FACS enrichment of paired
D10A Cas9 nickases will significantly facilitate efficient gene editing with minimized off-target effects
[28]. Further modifications showed that hRad51–Cas9 nickase, generated by hRad51 mutants fusion with Cas9
(D10A) nickase (RDN), mediates HDR without double-stranded breaks, which results in higher HDR:indel ratios
and lower off-target activity than Cas9 nuclease [29].
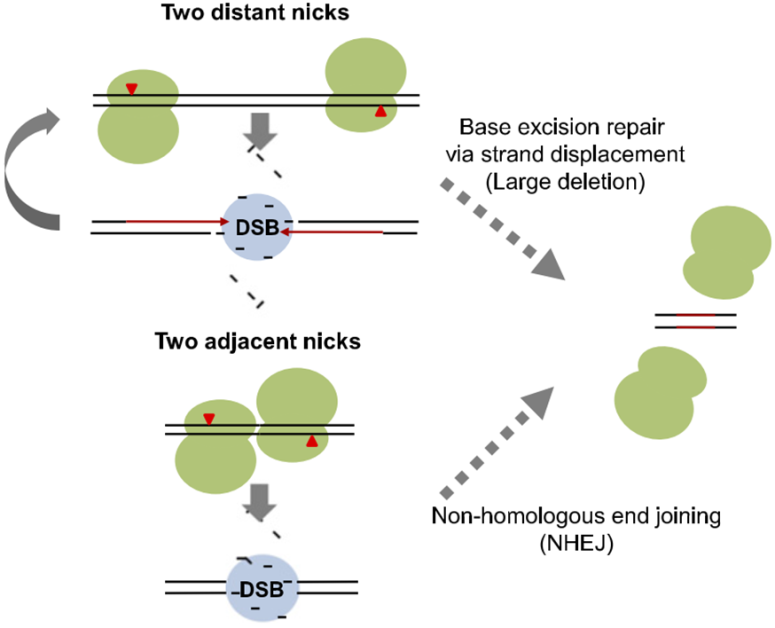
Figure 3. A schematic model of paired Cas9 nickase-mediated chromosomal deletions. Newly synthesized DNA
strands are shown in red [25].
c. dCas9 application: I) dCas9 activation system: MPH, VP64; II) dCas9 repression system
The nuclease-deactivated Cas9 variant, dCas9, was generated by introducing two mutations (D10A and H840A)
(Figure. 2A), resulting in no catalytic activity in both RuvC domain and HNH domain [30]. With great DNA
binding activity, dCas9 has been widely applied to regulate gene expression in combination with
transcription activator or repressor [14,31] (Table 3). (I) dCas9 activation system: transcription activator
domains (VP64, p65, MPH) or epigenetic activation modifications (p300, CBP etc.), have been fused to dCas9
to activate gene expression [32-35]; (II) dCas9 repression system: transcription repressor domains (KRAB) or
epigenetic repression modifications (LSD1, DNMT3A etc.), have been fused to down-regulate the expression of
target genes [34,36-38]. Besides the direct effector fusion manner, functional scaffolds (such as SunTag)
can be incorporated into dCas9/sgRNA complex to recruit effectors to affect transcription [36,39].
Spatiotemporal control in dCas9/sgRNA-induced effector recruitment was also achieved upon chemical or light
induction [40,41].
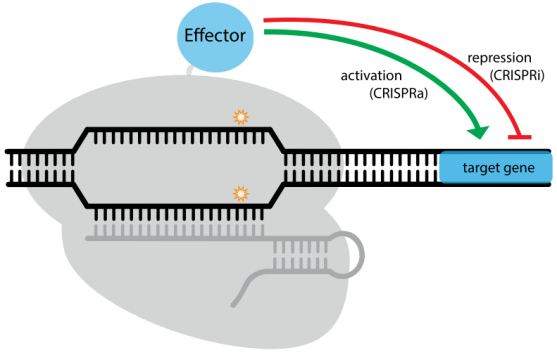
Figure 4. A modular RNA-guided genome regulation platform: CRISPR/dCas9 system. Targeted by sgRNA, dCas9
can mediate transcription activation or repression via genetically fusion with specific effectors
[42].
| Table 1. Effectors in dCas9 systems | ||
| Effector | Transcription regulation | |
| Activator | VP64 | transcription activation |
| Activator | VP48 | transcription activation |
| Activator | VP120 | transcription activation |
| Activator | p65 | transcription activation |
| Activator | MPH (MS2-P65-HSF1) | transcription activation |
| Activator | P65-HSF1 | transcription activation |
| Activator | VP64-p65-Rta | transcription activation |
| Repressor | KRAB | transcription repression |
| Repressor | SID4x | transcription repression |
| Repressor | Mxi1 | transcription repression |
| Histone acetylation | p300 | transcription activation |
| Histone acetylation | CBP | transcription activation |
| Histone demethylation | LSD1 | transcription repression |
| DNA methylation | DNMT3A | transcription repression |
| DNA demethylation | TET1 | transcription activation |
2) Other CRISPR system: Cas13, Cpf1, etc.
The traditional CRISPR/Cas9 system requires specific PAM sequence, which might be an obstacle to perform
gene editing in some cases. Moreover, the off-target effect and cytotoxicity also limit the application of
CRISPR/Cas9 system. To overcome these difficulties, researchers successfully screened several other tools to
perfect the gene editing system, such as CRISPR/Cas13 and CRISPR/Cpf1. etc.
a. Cas13
Cas13 family, containing Cas13a (also known as C2c2), Cas13b and Cas13c [43], belongs to class 2 type VI and
have two higher eukaryotes and prokaryotes nucleotide-binding (HEPN) endoRNase domains to mediate precise
RNA cleavage in targets with protospacer flanking sites [44,45]. Cas13 enzymes are programmable and have
been applied for nucleic acid detection as well as RNA knockdown and transcript tracking [46,47]. Combined
with base editor, such as ADAR2 (adenosine deaminase acting on RNA type 2), catalytically inactive Cas13
(dCas13) have been used as guidance with no strict sequence constraints to direct RNA base editing, which is
a promising platform with great applicability for both basic research and clinical therapy [48].
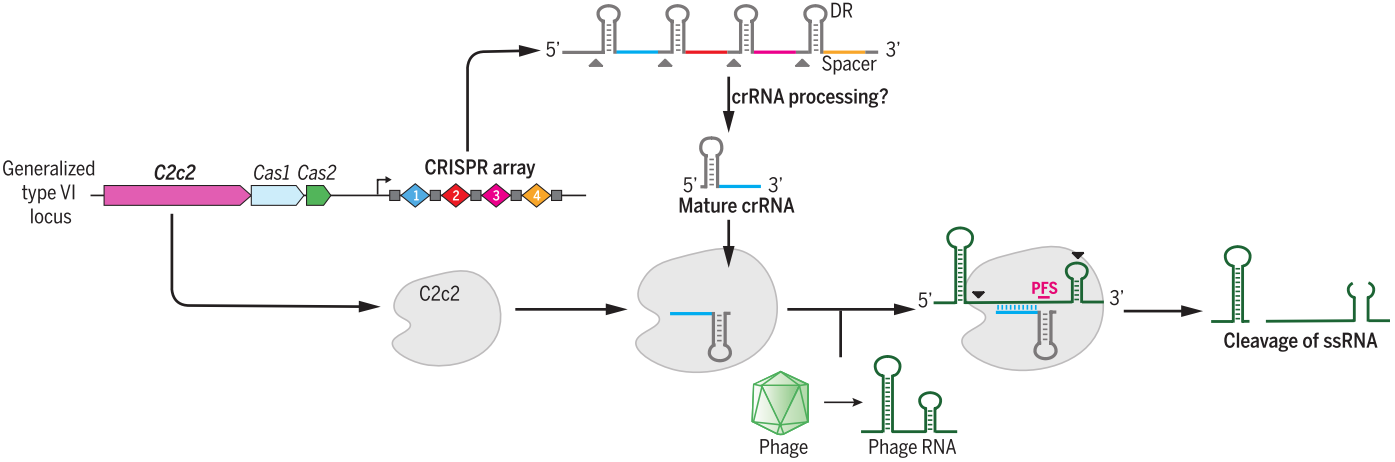 Figure 5. C2c2 (Cas13a) is a crRNA-guided RNase to cleavage of exogenous RNAs. Guided by crRNA, C2c2
mediates RNA CLEAVAGE without the need of sequence complementarity to crRNA guide sequence
[44].
b. Cpf1
Distinct with Cas9 enzyme, Cpf1 is from class 2 type V CRISPR system (Prevotella and Francisella 1) and
mediates DNA cleavage with three features. 1) The guiding sequence is processed into mature crRNA with no
requirement of the tracrRNA. 2) Guided by mature crRNA, Cpf1 efficiently cut the DNA with a short T-rich
protospacer-adjacent motif (PAM), not the G-rich PAM in Cas9 system. 3) Cpf1-crRNA system generates a
staggered DNA double-stranded break with a 4 or 5-nt 5’ overhang [49]. These features greatly expand the
application of CRISPR system [50].
Figure 5. C2c2 (Cas13a) is a crRNA-guided RNase to cleavage of exogenous RNAs. Guided by crRNA, C2c2
mediates RNA CLEAVAGE without the need of sequence complementarity to crRNA guide sequence
[44].
b. Cpf1
Distinct with Cas9 enzyme, Cpf1 is from class 2 type V CRISPR system (Prevotella and Francisella 1) and
mediates DNA cleavage with three features. 1) The guiding sequence is processed into mature crRNA with no
requirement of the tracrRNA. 2) Guided by mature crRNA, Cpf1 efficiently cut the DNA with a short T-rich
protospacer-adjacent motif (PAM), not the G-rich PAM in Cas9 system. 3) Cpf1-crRNA system generates a
staggered DNA double-stranded break with a 4 or 5-nt 5’ overhang [49]. These features greatly expand the
application of CRISPR system [50].
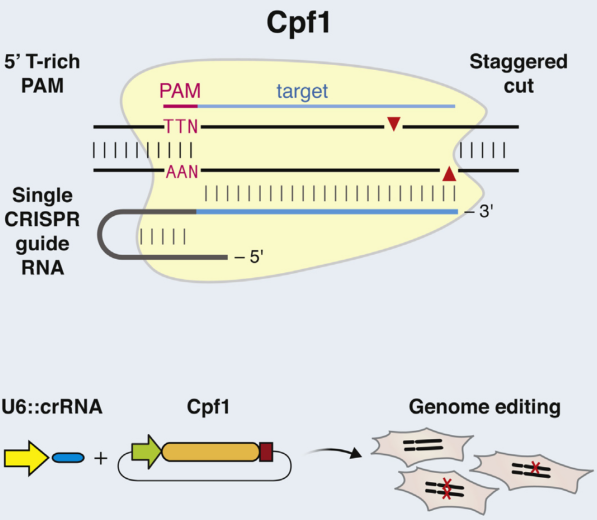
Figure 6. Cpf1 is a RNA-guided DNA nuclease cleaving target DNA with a T-rich protospacer-adjacent motif
via a staggered DNA double-stranded break.
CRISPR/Cas9 delivery system
CRISPR/Cas9 system can be delivered in vitro by the non-viral reagents (such as polymer, liposome, nano-particle, electroporation) or viral vectors (such as lentivirus, adenovirus and AAV) and in vivo by adenovirus or AAV vectors.
1)In vitro non-viral vectors CRISPR-Cas9 delivery system
In vitro non-viral vectors CRISPR/Cas9 delivery system can be mediated by non-viral reagents, such as polymer [51], liposome [52], nano-particle [53], electroporation [54]. The characteristics of these methods are summarized in the following table 2. For more detailed information about the transfection reagents, you can refer to the website: www.genemedi.net/i/transfection-in-vitro-in-vivo.
|
Table 2 - Comparison between polymer, liposome, nano-particle. |
||||
|
Comparison |
polymer |
liposome |
nano-particle |
electroporation |
|
Principle |
endocytosis |
endocytosis |
membrane fusion/impale cells |
transient increase in the permeability of cell membrane |
|
Integration |
No |
No |
No |
No |
|
Time to peak expression |
48h-72h |
48h-72h |
24h-48h |
48h-72h |
|
Sutainable time |
transient expression |
transient expression |
transient expression |
transient expression |
|
Cell Type |
a number of cell types |
adherent and suspension cell lines |
Almost all the cells |
not suitable for sensitive cell types |
|
Particle diametre |
75 to 520 nm |
50-200nm |
100-300nm |
—— |
|
Animal experiment |
low effiency |
low effiency |
target delivery |
target delivery |
|
Cytotoxicity |
high |
low |
non-toxic |
—— |
|
Immune Response |
No |
No |
No |
No |
|
Efficiency |
low effiency (<10%) |
depend on cell type |
high efficiency |
high efficiency |
|
Price |
inexpensive |
medium |
expensive |
most expensive (electroporation device) |
2) In intro viral vectors CRISPR-Cas9 delivery system
a. lentiviral With the ability to mediate efficient transfection and long-term expression of exogenous genes in both dividing and non-dividing cells, lentiviral is a good vector to deliver CRISPR system [55,56]. More useful information of lentivirus can be found on this website: www.genemedi.net/i/lentivirus-packaging. b. adenoviral Based on human adenovirus type 5 (Ad5), recombinant adenovirus (Ad) a replication-defective adenoviral vector system, is widely used for CRISPR-Cas9 system in most cell types [57-59]. Genemedi got a rich experience in adenovirus packaging, you could find more information on www.genemedi.net/i/adenovirus-packaging
3) In vivo CRISPR-Cas9 delivery system-AAV-SaCas9
With mild immunogenicity, adeno-associated viruses (AAV) have superior biosafety rating and broad range of infectivity and mediate long-term and stable expression of CRISPR system in vivo. Considering the loading capacity of AAV (4.7kb), SaCas9 variant has been identified, referred to as KKH SaCas9, displaying robust genome editing activities at target sites with NNNRRT PAMs and showing comparable off-target effects between wild-type and KKH SaCas9 [60]. Combining CRISPR/SaCas9 system and AAV vector, AAV-SaCas9 has been widely applied for gene editing in vivo [61-63]. Genemedi is good at AAV production, please find more information on this website: www.genemedi.net/i/aav-packaging.
|
Table 3 - Comparison between Retrovirus, Lentivirus, Adenovirus and AAV vectors. |
||||
|
Comparison |
Retrovirus |
Lentivirus |
Adenovirus |
AAV |
|
Genome |
ss RNA |
ss RNA |
ds DNA |
ss DNA |
|
Integration |
Yes |
Yes |
No |
No |
|
Packaging Capacity |
3kb |
4kb |
5.5kb |
2kb |
|
Time to peak expression |
72h |
72h |
36h-72h |
cell: 7 days; animals: 2 weeks |
|
Sutainable time |
about 3 weeks |
stable expression |
transient expression |
> 6 months |
|
Cell Type |
Most Dividing |
Most Dividing/Non-Dividing Cells |
Most Dividing/Non-Dividing Cells |
Most Dividing/Non-Dividing Cells |
|
Titeration |
10^7 TU/ml |
10^8 TU/ml |
10^11 PFU/ml |
10^12 v.g./ml |
|
Animal experiment |
suitable |
low efficiency |
lowest efficiency |
most suitable |
|
Immune Response |
high |
medium |
medium |
ultra-low |
Protocol of CRISPR /Cas9 mediated gene knockout in vitro and in vivo
Genemedi has launched a comprehensive AAV packaging service combined with CRISPR/Cas9, the versatile genome-editing platform. The followings are some protocols of CRISPR /Cas9 mediated gene knockout in vitro and in vivo.
For AAV CRISPR/Cas9 service, please visit:
https://www.genemedi.net/i/aav-sacas9-packaging
For lentivirus CRISPR/Cas9 service, please visit:
https://www.genemedi.net/i/crispr-cas9-knockout-lentivirus-production-service
For Adenovirus CRISPR/Cas9 service, please visit:
https://www.genemedi.net/i/adenovirus-cas9-packaging
CRISPR/Cas9 AAV Production-User Manual
https://www.genemedi.net/pdf/Genemedi-AAV-Sa Cas9%20User%20Manual.pdf
Recombinant Adenovirus-CRISPR/Cas9 Knockout System-User Manual
https://www.genemedi.net/pdf/Genemedi-Adenovirus-crispr%20User%20Manual.pdf
Recombinant Lentivirus-CRISPR/Cas9 Knockout System-User Manual
https://www.genemedi.net/pdf/Genemedi-Lentivirus-crispr%20User%20Manual.pdf
gRNA design and validation
Cas9-gRNA design principles:
1) The PAM sequence that SpCas9 recognizes is NGG (AGG, TGG, CGG, GGG), while SaCas9 recognizes NNGRRT (NNGAAT, NNGAGT, NNGGAT, NNGGGT) or NNGRRN. For sgRNA, the length is about 21 or 22 nucleotides.
2) For sgRNA, the GC content in 40%~60% is better.
3) If the sgRNA is driven by U6 or T7 promoter, the 5’ end of sgRNA can be designed as G or GG to improve transcription efficiency, which should be considered.
4) The binding site of sgRNA should be as close to the coding region in the downstream of ATG as possible to induce frameshift mutation, the first or second exon is better.
5) SNPs should be checked in the binding site of sgRNA.
6) The distance between paired sgRNA should be taken into consideration before designing paired-gRNAs, if using Cas9 single nickase.
7) Whole genome off-target effect analysis is suggested. At least 5 bases can be allowed for the base mismatch and whether the off target is located in the gene encoding regions need to be confirmed. What’s more, base insertion or deletion in off targets should be detected.
To date, there exist so many CRISPR/Cas9 design tools, and some common tools are list in table 1.
|
Name |
Web |
Affiliation |
|
CRISPR design |
http://crispr.mit.edu/ |
Massachusetts Institute of Technology |
|
CRISPR design tool |
The Broad Institute of Harvard and MIT |
|
|
Cas-OFFinder |
http://www.rgenome.net |
Harvard Medical School |
|
GPP sgRNA Designer |
https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
The Broad Institute of Harvard and MIT |
1)SpCas9 gRNA design, database and validation
Based on the above principles, SpCas9-gRNA can be designed on the following website [64]: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. Choose the CRISPR enzyme type “SpyoCas9 (NGG)”, then input transcript IDs, Gene IDs/Symbols, raw DNA sequence or upload a list of transcript IDs, Gene IDs/Symbols, a FASTA file of DNA sequences. Click on the “submit” button, and you can get and download the gRNA sequence from the following website.
Synthesize the SpCas9 gRNA sequence and its anti-sense sequence, anneal the sequences into a double chain and clone into pLv-SpCas9 vector or pAd-SpCas9 vector, which is confirmed by Sanger sequencing.
Deliver the pLv-SpCas9 vector or pAd-SpCas9 vector containing target sgRNA sequences into cells, and verify knockout efficiency.
2)SaCas9 gRNA design, database and validation
Based on the above principles, SaCas9-gRNA can be designed on the following website [64]: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. Choose the CRISPR enzyme type “SaurCas9 (NNGRR)”, then input transcript IDs, Gene IDs/Symbols, raw DNA sequence or upload a list of transcript IDs, Gene IDs/Symbols, a FASTA file of DNA sequences. Click on the “submit” button, and you can get and download the gRNA sequence from the following website.
Synthesize the SaCas9 gRNA sequence and its anti-sense sequence, anneal the sequences into a double chain and clone into pAAV-SaCas9 vector, which is confirmed by Sanger sequencing.
Deliver the pAAV-SaCas9 vector containing target sgRNA sequences into cells, and verify knockout efficiency.
3)Summary of approaches for gRNA validation
The cutting efficiency of Cas9 nuclease is mainly affected gRNA target efficiency, which can be validated by
T7 endonuclease I (T7EI) assay, Sanger sequencing and Western blot.
a.T7 endonuclease I
(T7EI) assay
T7 Endonuclease I recognizes and cleaves non-perfectly matched DNA, which is a
simple and cheap method to determine genome targeting efficiency of CRISPR/Cas9 system. First, extract the
genomic DNA of cells whose genomes were targeted by Cas9. Second, perform PCR experiments to amplify target
DNA. Third, anneal the PCR products and digest them with T7 Endonuclease I. Fourth, analyze the fragments
with agarose gel to determine the efficiency of gRNA targeting.
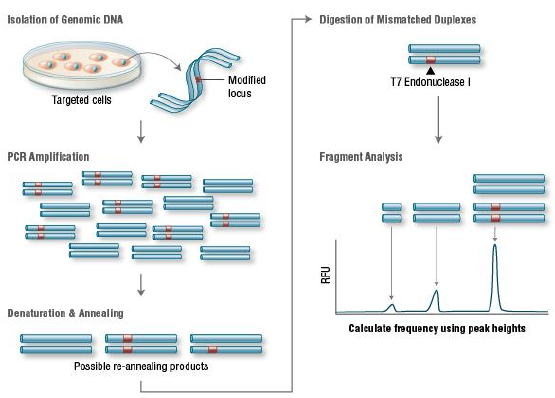
Figure 7. Principle of T7E1 assay for knockout clones
b.Sanger sequencing assay
Sanger sequencing is a direct and precise way to examine the
target efficiency of gRNA. First, extract the genomic DNA of cells whose genomes were targeted by Cas9.
Second, perform PCR experiments to amplify target DNA. Third, verify the sequence of target genome by Sanger
sequencing and then calculate the cleavage efficiency.
c.Western blot (WB) assay
Western blot is also simple, easy but no so precise assay
for the determination of gRNA targeting efficiency. First, extract the whole cell protein of cells whose
genomes were targeted by Cas9. Second, perform Western blot experiments to detect the expression levels of
target protein. Third, confirm the efficiency of gRNA by analyzing the WB results.
4)How to detect the off-target?
Although CRISPR/Cas9 system is a versatile gene editing technology that is widely applied in the study of basic science and translational research, high frequency of off-target activity is really a restriction factor for its therapeutic and clinical applications. So far, several methods have been used to determine the off-targets with different advantages and disadvantages [65].
|
Table 5. Common CRISPR/Cas9 design tools [65] |
|||
|
Method |
Advantage |
Disadvantage |
References |
|
T7E1 assay |
Simple |
Poor sensitivity, not cost-effective |
|
|
Deep sequencing |
Precise |
Biased, misses potential off-target sites elsewhere in the genome |
|
|
In silico prediction |
Predicts some off-target mutation sites |
Fails to predict bona-fide off-target sites |
|
|
ChIP-seq |
Unbiased detection of Cas9 binding sites genome-wide |
Most off-target DNA-binding sites recognized by dCas9 are not cleaved at all by Cas9 in cells |
[71-74] |
|
GUIDE-seq |
Unbiased, sensitive (0.1%), qualitative translocations, identifies breakpoint hotspots |
False negatives present, limited by chromatin accessibility |
[75] |
|
HTGTS |
Identifies translocations |
False negatives present, limited by chromatin accessibility |
[73] |
|
IDLV |
Programmable, sensitive (1%) |
Many bona-fide off-target sites cannot be captured |
[76] |
|
Digenome-seq |
Sensitive (0.1% or lower), unbiased and cost-effective |
Not widely used |
[77] |
|
FISH |
Quick |
Less precise |
[78] |
ChIP-seq, Chromatin immunoprecipitation followed by
massively parallel DNA sequencing
GUIDE-seq, Genome-wide unbiased identification of DSBs enabled by
sequencing
HTGTS, high-throughput, genome-wide, translocation sequencing
IDLV,
integrase-defective lentiviral vectors
FISH, fluorescence in situ hybridization
5)How to minimize off-target?
a. Alter the sgRNA sequence. Truncation of the 3′ end of sgRNA, shortening the region complementary to the
target site at the 5′ end of the sgRNA by as many as 3 nt (tru-gRNA) or addition of two guanine nucleotides
to the 5′ end of the sgRNA improves target specificity [79].
b. Try to optimize the design of Cas9 and sgRNA design with design tools or directed evolution
[64,80].
c. Purified Cas9 ribonucleoprotein works more immediately than plasmids containing Cas9 gene
sequence and can be degraded rapidly in cells, which may help minimize the off-target frequency [81,82].
d. Strategies that restrict the transcription and translation of Cas9 may help minimize
off-target effects [83].
e. Replace the wild-type Cas9 nuclease with D10 mutant Cas9 nickase and paired with two sgRNAs
that each cleaves only one strand [28].
f. Improve DNA cleavage specificity using fusions of dCas9 with FokI nuclease domain (fCas9)
(Figure 6) [84,85].
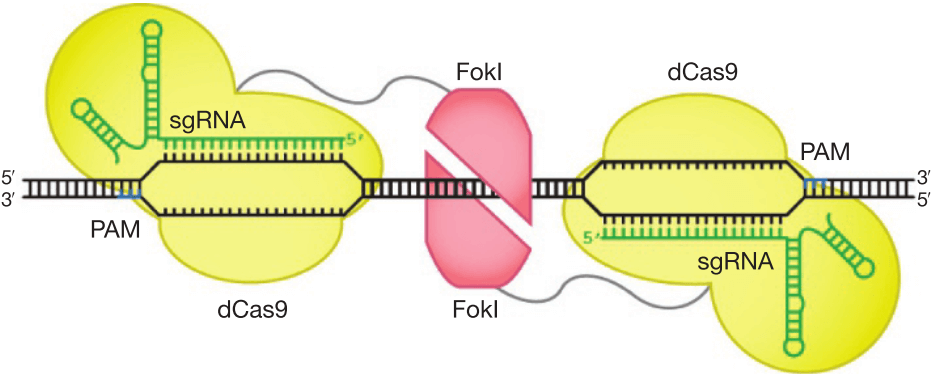
Figure 8. Architectures of Cas9 and FokI-dCas9 fusion variants. Two monomers of FokI nuclease (red) fused to dCas9-sgRNAs as shown in figure cleavage nucleic acids within the target locus. The catalytically active FokI nuclease dimer can only be assembled by adjacently bound by two FokI-dCas9 monomers to trigger dsDNA cleavage [84].
Gene editing therapy
1)Gene editing therapy vs gene replacement therapy
Gene therapy, to be brief, is delivering corrective gene materials into cells to treat or alleviate the symptom of disease [86], so far, including gene editing therapy mediated by CRISPR/Cas sytem, ZFN or TALEN etc., and gene replacement, such as lentivirus/adenovirus/AAV-mediated gene expression [20]. Gene editing therapy via CRISPR technology is to correct the pathogenic mutation into non-pathogenic ones, such as gene silence or disruption by CRISPR mediated knockout, which has been widely used in the therapy of hemophilia B [19], Huntington's disease [87], Parkinson's Disease [88], hematologic diseases, infectious diseases and malignant tumor [89]. Gene replacement therapy by lentivirus/adenovirus/AAV is to replace the pathogenic gene with its corrective type, which is widely applied in treating lipoprotein lipase deficiency (LPLD) [90], spinal muscular atrophy (SMA) [91], retinal dystrophy [92,93], cystic fibrosis [94,95] muscular dystrophies [96]. Combined CRISPR with virus vectors, AAV-SaCas9 or lentivirus-Cas9 has been used for gene therapy of Hemophilia A [61], Usher Syndrome [97], Intervertebral Disc Degeneration [98] etc. Click here for Pipelines landscapes of gene replacement therapy and gene editing therapy of different companies..
2) Landscape of gene editing therapy: companies and pipelines
As a powerful and versatile platform, gene editing therapy has been led into a new era. Besides basic research, great progress has been made in the clinical studied based on CRISPR. Several companies have been operated to develop transformative genomic medicines for the treatment a range of genetically addressable diseases. Founded by Zhangfeng, Jennifer A. Doudna, George Church, J. Keith Joung and David R. Liu in 2013, Editas Medicine, Inc. develops several gene medicines for disease therapy, such as EDIT-101 for the treatment of Leber Congenital Amaurosis type 10, which has entered into the phase I/II dose escalation study. Founded in Swiss in 2013, CRISPR Therapeutics has developed gene medicine CTX001 for the therapy of anemia, showing promising data in the phase I/II clinical study. Operated by Zhangfeng, David R. Liu and J. Keith Joung in 2018, Beam Therapeutics focuses on treating genetic diseases caused by point mutations with CRISPR-mediated single base editing technique. Click here for Pipelines landscapes of gene replacement therapy and gene editing therapy of different companies..
Reference
1.
Sapranauskas R, G Gasiunas, C Fremaux, R Barrangou, P Horvath and V Siksnys. (2011).
The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids
Res 39:9275-82.
2.
Barrangou R. (2015). Diversity of CRISPR-Cas immune systems and molecular machines.
Genome Biol 16:247.
3.
Wiedenheft B, E van Duijn, JB Bultema, SP Waghmare, K Zhou, A Barendregt, W Westphal,
AJ Heck, EJ Boekema, MJ Dickman and JA Doudna. (2011). RNA-guided complex from a bacterial immune system
enhances target recognition through seed sequence interactions. Proc Natl Acad Sci U S A
108:10092-7.
4.
Bhaya D, M Davison and R Barrangou. (2011). CRISPR-Cas systems in bacteria and
archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273-97.
5.
Terns MP and RM Terns. (2011). CRISPR-based adaptive immune systems. Curr Opin
Microbiol 14:321-7.
6.
Kleinstiver BP, MS Prew, SQ Tsai, VV Topkar, NT Nguyen, Z Zheng, AP Gonzales, Z Li,
RT Peterson, JR Yeh, MJ Aryee and JK Joung. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM
specificities. Nature 523:481-5.
7.
Jinek M, K Chylinski, I Fonfara, M Hauer, JA Doudna and E Charpentier. (2012). A
programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816-21.
8.
Ablain J, EM Durand, S Yang, Y Zhou and LI Zon. (2015). A CRISPR/Cas9 vector system
for tissue-specific gene disruption in zebrafish. Dev Cell 32:756-64.
9.
Guo T, YL Feng, JJ Xiao, Q Liu, XN Sun, JF Xiang, N Kong, SC Liu, GQ Chen, Y Wang, MM
Dong, Z Cai, H Lin, XJ Cai and AY Xie. (2018). Harnessing accurate non-homologous end joining for
efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol 19:170.
10.
Wang B, K Li, A Wang, M Reiser, T Saunders, RF Lockey and JW Wang. (2015). Highly
efficient CRISPR/HDR-mediated knock-in for mouse embryonic stem cells and zygotes. Biotechniques
59:201-2, 204, 206-8.
11.
Zhang JP, XL Li, GH Li, W Chen, C Arakaki, GD Botimer, D Baylink, L Zhang, W Wen, YW
Fu, J Xu, N Chun, W Yuan, T Cheng and XB Zhang. (2017). Efficient precise knockin with a double cut HDR
donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol 18:35.
12.
Men K, X Duan, Z He, Y Yang, S Yao and Y Wei. (2017). CRISPR/Cas9-mediated correction
of human genetic disease. Sci China Life Sci 60:447-457.
13.
Panier S and SJ Boulton. (2014). Double-strand break repair: 53BP1 comes into focus.
Nat Rev Mol Cell Biol 15:7-18.
14.
Yang H, H Wang, CS Shivalila, AW Cheng, L Shi and R Jaenisch. (2013). One-step
generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering.
Cell 154:1370-9.
15.
Wang H, H Yang, CS Shivalila, MM Dawlaty, AW Cheng, F Zhang and R Jaenisch. (2013).
One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome
engineering. Cell 153:910-8.
16.
Mali P, L Yang, KM Esvelt, J Aach, M Guell, JE DiCarlo, JE Norville and GM Church.
(2013). RNA-guided human genome engineering via Cas9. Science 339:823-6.
17.
Maruyama T, SK Dougan, MC Truttmann, AM Bilate, JR Ingram and HL Ploegh. (2015).
Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end
joining. Nat Biotechnol 33:538-42.
18.
Roman-Rodriguez FJ, L Ugalde, L Alvarez, B Diez, MJ Ramirez, C Risueno, M Corton, M
Bogliolo, S Bernal, F March, C Ayuso, H Hanenberg, J Sevilla, S Rodriguez-Perales, R Torres-Ruiz, J
Surralles, JA Bueren and P Rio. (2019). NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks
Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell 25:607-621
e7.
19.
Huai C, C Jia, R Sun, P Xu, T Min, Q Wang, C Zheng, H Chen and D Lu. (2017).
CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice.
Hum Genet 136:875-883.
20.
Lamsfus-Calle A, A Daniel-Moreno, G Urena-Bailen, J Raju, JS Antony, R Handgretinger
and M Mezger. (2019). Hematopoietic stem cell gene therapy: The optimal use of lentivirus and gene
editing approaches. Blood Rev:100641.
21.
Tran NT, T Sommermann, R Graf, J Trombke, J Pempe, K Petsch, R Kuhn, K Rajewsky and
VT Chu. (2019). Efficient CRISPR/Cas9-Mediated Gene Knockin in Mouse Hematopoietic Stem and Progenitor
Cells. Cell Rep 28:3510-3522 e5.
22.
Chen S, S Sun, D Moonen, C Lee, AY Lee, DV Schaffer and L He. (2019). CRISPR-READI:
Efficient Generation of Knockin Mice by CRISPR RNP Electroporation and AAV Donor Infection. Cell Rep
27:3780-3789 e4.
23.
Shalem O, NE Sanjana, E Hartenian, X Shi, DA Scott, T Mikkelson, D Heckl, BL Ebert,
DE Root, JG Doench and F Zhang. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells.
Science 343:84-87.
24.
San Filippo J, P Sung and H Klein. (2008). Mechanism of eukaryotic homologous
recombination. Annu Rev Biochem 77:229-57.
25.
Cho SW, S Kim, Y Kim, J Kweon, HS Kim, S Bae and JS Kim. (2014). Analysis of
off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res
24:132-41.
26.
Chiang TW, C le Sage, D Larrieu, M Demir and SP Jackson. (2016). CRISPR-Cas9(D10A)
nickase-based genotypic and phenotypic screening to enhance genome editing. Sci Rep 6:24356.
27.
Gopalappa R, B Suresh, S Ramakrishna and HH Kim. (2018). Paired D10A Cas9 nickases
are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res
46:e71.
28.
Gopalappa R, M Song, AP Chandrasekaran, S Das, S Haq, HC Koh and S Ramakrishna.
(2018). Efficient genome editing by FACS enrichment of paired D10A Cas9 nickases coupled with
fluorescent proteins. Arch Pharm Res 41:911-920.
29.
Rees HA, WH Yeh and DR Liu. (2019). Development of hRad51-Cas9 nickase fusions that
mediate HDR without double-stranded breaks. Nat Commun 10:2212.
30.
Bikard D, W Jiang, P Samai, A Hochschild, F Zhang and LA Marraffini. (2013).
Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas
system. Nucleic Acids Res 41:7429-37.
31.
Cheng AW, H Wang, H Yang, L Shi, Y Katz, TW Theunissen, S Rangarajan, CS Shivalila,
DB Dadon and R Jaenisch. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided
transcriptional activator system. Cell Res 23:1163-71.
32.
Chavez A, J Scheiman, S Vora, BW Pruitt, M Tuttle, PRI E, S Lin, S Kiani, CD Guzman,
DJ Wiegand, D Ter-Ovanesyan, JL Braff, N Davidsohn, BE Housden, N Perrimon, R Weiss, J Aach, JJ Collins
and GM Church. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat Methods
12:326-8.
33.
Gilbert LA, MH Larson, L Morsut, Z Liu, GA Brar, SE Torres, N Stern-Ginossar, O
Brandman, EH Whitehead, JA Doudna, WA Lim, JS Weissman and LS Qi. (2013). CRISPR-mediated modular
RNA-guided regulation of transcription in eukaryotes. Cell 154:442-51.
34.
Hilton IB, AM D'Ippolito, CM Vockley, PI Thakore, GE Crawford, TE Reddy and CA
Gersbach. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from
promoters and enhancers. Nat Biotechnol 33:510-7.
35.
Cheng AW, N Jillette, P Lee, D Plaskon, Y Fujiwara, W Wang, A Taghbalout and H Wang.
(2016). Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell
Res 26:254-7.
36.
Gilbert LA, MA Horlbeck, B Adamson, JE Villalta, Y Chen, EH Whitehead, C Guimaraes, B
Panning, HL Ploegh, MC Bassik, LS Qi, M Kampmann and JS Weissman. (2014). Genome-Scale CRISPR-Mediated
Control of Gene Repression and Activation. Cell 159:647-61.
37.
Kearns NA, H Pham, B Tabak, RM Genga, NJ Silverstein, M Garber and R Maehr. (2015).
Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods
12:401-403.
38.
Vojta A, P Dobrinic, V Tadic, L Bockor, P Korac, B Julg, M Klasic and V Zoldos.
(2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res
44:5615-28.
39.
Tanenbaum ME, LA Gilbert, LS Qi, JS Weissman and RD Vale. (2014). A protein-tagging
system for signal amplification in gene expression and fluorescence imaging. Cell 159:635-46.
40.
Konermann S, MD Brigham, A Trevino, PD Hsu, M Heidenreich, L Cong, RJ Platt, DA
Scott, GM Church and F Zhang. (2013). Optical control of mammalian endogenous transcription and
epigenetic states. Nature 500:472-476.
41.
Nihongaki Y, S Yamamoto, F Kawano, H Suzuki and M Sato. (2015). CRISPR-Cas9-based
photoactivatable transcription system. Chem Biol 22:169-74.
42.
Brocken DJW, M Tark-Dame and RT Dame. (2018). dCas9: A Versatile Tool for Epigenome
Editing. Curr Issues Mol Biol 26:15-32.
43.
Shmakov S, A Smargon, D Scott, D Cox, N Pyzocha, W Yan, OO Abudayyeh, JS Gootenberg,
KS Makarova, YI Wolf, K Severinov, F Zhang and EV Koonin. (2017). Diversity and evolution of class 2
CRISPR-Cas systems. Nat Rev Microbiol 15:169-182.
44.
Abudayyeh OO, JS Gootenberg, S Konermann, J Joung, IM Slaymaker, DB Cox, S Shmakov,
KS Makarova, E Semenova, L Minakhin, K Severinov, A Regev, ES Lander, EV Koonin and F Zhang. (2016).
C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science
353:aaf5573.
45.
East-Seletsky A, MR O'Connell, SC Knight, D Burstein, JH Cate, R Tjian and JA Doudna.
(2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection.
Nature 538:270-273.
46.
Gootenberg JS, OO Abudayyeh, JW Lee, P Essletzbichler, AJ Dy, J Joung, V Verdine, N
Donghia, NM Daringer, CA Freije, C Myhrvold, RP Bhattacharyya, J Livny, A Regev, EV Koonin, DT Hung, PC
Sabeti, JJ Collins and F Zhang. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science
356:438-442.
47.
Abudayyeh OO, JS Gootenberg, P Essletzbichler, S Han, J Joung, JJ Belanto, V Verdine,
DBT Cox, MJ Kellner, A Regev, ES Lander, DF Voytas, AY Ting and F Zhang. (2017). RNA targeting with
CRISPR-Cas13. Nature 550:280-284.
48.
Cox DBT, JS Gootenberg, OO Abudayyeh, B Franklin, MJ Kellner, J Joung and F Zhang.
(2017). RNA editing with CRISPR-Cas13. Science 358:1019-1027.
49.
Zetsche B, JS Gootenberg, OO Abudayyeh, IM Slaymaker, KS Makarova, P Essletzbichler,
SE Volz, J Joung, J van der Oost, A Regev, EV Koonin and F Zhang. (2015). Cpf1 is a single RNA-guided
endonuclease of a class 2 CRISPR-Cas system. Cell 163:759-71.
50.
Fagerlund RD, RH Staals and PC Fineran. (2015). The Cpf1 CRISPR-Cas protein expands
genome-editing tools. Genome Biol 16:251.
51.
Kang YK, K Kwon, JS Ryu, HN Lee, C Park and HJ Chung. (2017). Nonviral Genome Editing
Based on a Polymer-Derivatized CRISPR Nanocomplex for Targeting Bacterial Pathogens and Antibiotic
Resistance. Bioconjug Chem 28:957-967.
52.
Zhen S and X Li. (2019). Liposomal delivery of CRISPR/Cas9. Cancer Gene Ther.
53.
Chin JS, WH Chooi, H Wang, W Ong, KW Leong and SY Chew. (2019). Scaffold-mediated
non-viral delivery platform for CRISPR/Cas9-based genome editing. Acta Biomater 90:60-70.
54.
Yamashita S, Y Kogasaka, Y Hiradate, K Tanemura and Y Sendai. (2019). Suppression of
mosaic mutation by co-delivery of CRISPR associated protein 9 and three-prime repair exonuclease 2 into
porcine zygotes via electroporation. J Reprod Dev.
55.
Koike-Yusa H, Y Li, EP Tan, C Velasco-Herrera Mdel and K Yusa. (2014). Genome-wide
recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat
Biotechnol 32:267-73.
56.
McDade JR, NC Waxmonsky, LE Swanson and M Fan. (2016). Practical Considerations for
Using Pooled Lentiviral CRISPR Libraries. Curr Protoc Mol Biol 115:31 5 1-31 5 13.
57.
Lee J, J Ma and K Lee. (2019). Direct delivery of adenoviral CRISPR/Cas9 vector into
the blastoderm for generation of targeted gene knockout in quail. Proc Natl Acad Sci U S A
116:13288-13292.
58.
Romero Z, A Lomova, S Said, A Miggelbrink, CY Kuo, B Campo-Fernandez, MD Hoban, KE
Masiuk, DN Clark, J Long, JM Sanchez, M Velez, E Miyahira, R Zhang, D Brown, X Wang, YZ Kurmangaliyev,
RP Hollis and DB Kohn. (2019). Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem
Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol Ther 27:1389-1406.
59.
Lv P, X Liu, X Chen, C Liu, Y Zhang, C Chu, J Wang, X Wang, X Chen and G Liu. (2019).
Genetically Engineered Cell Membrane Nanovesicles for Oncolytic Adenovirus Delivery: A Versatile
Platform for Cancer Virotherapy. Nano Lett 19:2993-3001.
60.
Kleinstiver BP, MS Prew, SQ Tsai, NT Nguyen, VV Topkar, Z Zheng and JK Joung. (2015).
Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat
Biotechnol 33:1293-1298.
61.
Chen H, M Shi, A Gilam, Q Zheng, Y Zhang, I Afrikanova, J Li, Z Gluzman, R Jiang, LJ
Kong and RY Chen-Tsai. (2019). Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing
of human Factor VIII. Sci Rep 9:16838.
62.
Xu L, YS Lau, Y Gao, H Li and R Han. (2019). Life-Long AAV-Mediated CRISPR Genome
Editing in Dystrophic Heart Improves Cardiomyopathy without Causing Serious Lesions in mdx Mice. Mol
Ther 27:1407-1414.
63.
Pan X, L Philippen, SK Lahiri, C Lee, SH Park, TA Word, N Li, KE Jarrett, R Gupta, JO
Reynolds, J Lin, G Bao, WR Lagor and XHT Wehrens. (2018). In Vivo Ryr2 Editing Corrects
Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res 123:953-963.
64.
Doench JG, N Fusi, M Sullender, M Hegde, EW Vaimberg, KF Donovan, I Smith, Z Tothova,
C Wilen, R Orchard, HW Virgin, J Listgarten and DE Root. (2016). Optimized sgRNA design to maximize
activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184-191.
65.
Zhang XH, LY Tee, XG Wang, QS Huang and SH Yang. (2015). Off-target Effects in
CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids 4:e264.
66.
Cho SW, S Kim, JM Kim and JS Kim. (2013). Targeted genome engineering in human cells
with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230-2.
67.
Kim HJ, HJ Lee, H Kim, SW Cho and JS Kim. (2009). Targeted genome editing in human
cells with zinc finger nucleases constructed via modular assembly. Genome Res 19:1279-88.
68.
Cong L, FA Ran, D Cox, S Lin, R Barretto, N Habib, PD Hsu, X Wu, W Jiang, LA
Marraffini and F Zhang. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science
339:819-23.
69.
Ran FA, PD Hsu, J Wright, V Agarwala, DA Scott and F Zhang. (2013). Genome
engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281-2308.
70.
Heigwer F, G Kerr and M Boutros. (2014). E-CRISP: fast CRISPR target site
identification. Nat Methods 11:122-3.
71.
Wu X, DA Scott, AJ Kriz, AC Chiu, PD Hsu, DB Dadon, AW Cheng, AE Trevino, S
Konermann, S Chen, R Jaenisch, F Zhang and PA Sharp. (2014). Genome-wide binding of the CRISPR
endonuclease Cas9 in mammalian cells. Nat Biotechnol 32:670-6.
72.
Duan J, G Lu, Z Xie, M Lou, J Luo, L Guo and Y Zhang. (2014). Genome-wide
identification of CRISPR/Cas9 off-targets in human genome. Cell Res 24:1009-12.
73.
Kuscu C, S Arslan, R Singh, J Thorpe and M Adli. (2014). Genome-wide analysis reveals
characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32:677-83.
74.
Singh R, C Kuscu, A Quinlan, Y Qi and M Adli. (2015). Cas9-chromatin binding
information enables more accurate CRISPR off-target prediction. Nucleic Acids Res 43:e118.
75.
Tsai SQ, Z Zheng, NT Nguyen, M Liebers, VV Topkar, V Thapar, N Wyvekens, C Khayter,
AJ Iafrate, LP Le, MJ Aryee and JK Joung. (2015). GUIDE-seq enables genome-wide profiling of off-target
cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33:187-197.
76.
Wang X, Y Wang, X Wu, J Wang, Y Wang, Z Qiu, T Chang, H Huang, RJ Lin and JK Yee.
(2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective
lentiviral vectors. Nat Biotechnol 33:175-8.
77.
Kim D, S Bae, J Park, E Kim, S Kim, HR Yu, J Hwang, JI Kim and JS Kim. (2015).
Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods
12:237-43, 1 p following 243.
78.
Paulis M, A Castelli, M Lizier, L Susani, F Lucchini, A Villa and P Vezzoni. (2015).
A pre-screening FISH-based method to detect CRISPR/Cas9 off-targets in mouse embryonic stem cells. Sci
Rep 5:12327.
79.
Fu Y, JD Sander, D Reyon, VM Cascio and JK Joung. (2014). Improving CRISPR-Cas
nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279-284.
80.
Lee J, MH Jung, E Jeong and JK Lee. (2019). Using Sniper-Cas9 to Minimize Off-target
Effects of CRISPR-Cas9 Without the Loss of On-target Activity Via Directed Evolution. J Vis Exp.
81.
Kim S, D Kim, SW Cho, J Kim and JS Kim. (2014). Highly efficient RNA-guided genome
editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012-9.
82.
Ramakrishna S, AB Kwaku Dad, J Beloor, R Gopalappa, SK Lee and H Kim. (2014). Gene
disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res
24:1020-7.
83.
Shen CC, MN Hsu, CW Chang, MW Lin, JR Hwu, Y Tu and YC Hu. (2019). Synthetic switch
to minimize CRISPR off-target effects by self-restricting Cas9 transcription and translation. Nucleic
Acids Res 47:e13.
84.
Guilinger JP, DB Thompson and DR Liu. (2014). Fusion of catalytically inactive Cas9
to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577-582.
85.
Tsai SQ, N Wyvekens, C Khayter, JA Foden, V Thapar, D Reyon, MJ Goodwin, MJ Aryee and
JK Joung. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat
Biotechnol 32:569-76.
86.
Verma IM and N Somia. (1997). Gene therapy -- promises, problems and prospects.
Nature 389:239-42.
87.
Ekman FK, DS Ojala, MM Adil, PA Lopez, DV Schaffer and T Gaj. (2019).
CRISPR-Cas9-Mediated Genome Editing Increases Lifespan and Improves Motor Deficits in a Huntington's
Disease Mouse Model. Mol Ther Nucleic Acids 17:829-839.
88.
Safari F, G Hatam, AB Behbahani, V Rezaei, M Barekati-Mowahed, P Petramfar and F
Khademi. (2019). CRISPR System: A High-throughput Toolbox for Research and Treatment of Parkinson's
Disease. Cell Mol Neurobiol.
89.
Sun J, J Wang, D Zheng and X Hu. (2019). Advances in therapeutic application of
CRISPR-Cas9. Brief Funct Genomics.
90.
Kassner U, T Hollstein, T Grenkowitz, M Wuhle-Demuth, B Salewsky, I Demuth, M Dippel
and E Steinhagen-Thiessen. (2018). Gene Therapy in Lipoprotein Lipase Deficiency: Case Report on the
First Patient Treated with Alipogene Tiparvovec Under Daily Practice Conditions. Hum Gene Ther
29:520-527.
91.
Passini MA, J Bu, AM Richards, CM Treleaven, JA Sullivan, CR O'Riordan, A Scaria, AP
Kells, L Samaranch, W San Sebastian, T Federici, MS Fiandaca, NM Boulis, KS Bankiewicz, LS Shihabuddin
and SH Cheng. (2014). Translational fidelity of intrathecal delivery of self-complementary AAV9-survival
motor neuron 1 for spinal muscular atrophy. Hum Gene Ther 25:619-30.
92.
Stieger K and B Lorenz. (2014). [Specific gene therapy for hereditary retinal
dystrophies - an update]. Klin Monbl Augenheilkd 231:210-5.
93.
Trapani I, P Colella, A Sommella, C Iodice, G Cesi, S de Simone, E Marrocco, S Rossi,
M Giunti, A Palfi, GJ Farrar, R Polishchuk and A Auricchio. (2014). Effective delivery of large genes to
the retina by dual AAV vectors. EMBO Mol Med 6:194-211.
94.
Doi K and Y Takeuchi. (2015). Gene therapy using retrovirus vectors: vector
development and biosafety at clinical trials. Uirusu 65:27-36.
95.
Duncan GA, N Kim, Y Colon-Cortes, J Rodriguez, M Mazur, SE Birket, SM Rowe, NE West,
A Livraghi-Butrico, RC Boucher, J Hanes, G Aslanidi and JS Suk. (2018). An Adeno-Associated Viral Vector
Capable of Penetrating the Mucus Barrier to Inhaled Gene Therapy. Mol Ther Methods Clin Dev
9:296-304.
96.
Crudele JM and JS Chamberlain. (2019). AAV-based gene therapies for the muscular
dystrophies. Hum Mol Genet 28:R102-R107.
97.
Menghini M, J Cehajic-Kapetanovic, IH Yusuf and RE MacLaren. (2019). A novel
splice-site variant in CDH23 in a patient with Usher syndrome type 1. Ophthalmic Genet:1-4.
98.
Chen S, M Luo, H Kou, G Shang, Y Ji and H Liu. (2019). A Review of Gene Therapy
Delivery Systems for Intervertebral Disc Degeneration. Curr Pharm Biotechnol.
AAV Knowledge Base CRISPR/Cas9-gRNA system Knowledge Base |
Adenovirus Knowledge
Base Transfection Knowledge Base |
Lentivirus Knowledge Base Advanced cell therapy |
