




GeneMedi's protocol / procedure for the diagnostics application-ELISA
1 ELISA (enzyme-linked immunosorbent assay)
ELISA (enzyme-linked immunosorbent assay) is a method used to quantitatively detect an antigen within a sample.
An antigen is a toxin or other foreign substance, for example a flu virus or environmental contaminant, that causes the vertebrate immune system to mount a defensive response.
The range of potential antigens is vast, so ELISAs are used in many areas of research and testing to detect and quantify antigens in a wide variety of sample types. Cell lysates, blood samples, food items, and more can be analyzed for specific substances of interest using ELISAs.
There are four major types of ELISAs: direct, indirect, competitive and sandwich. Each type is described below with a diagram illustrating how the analytes and antibodies are bonded and used.
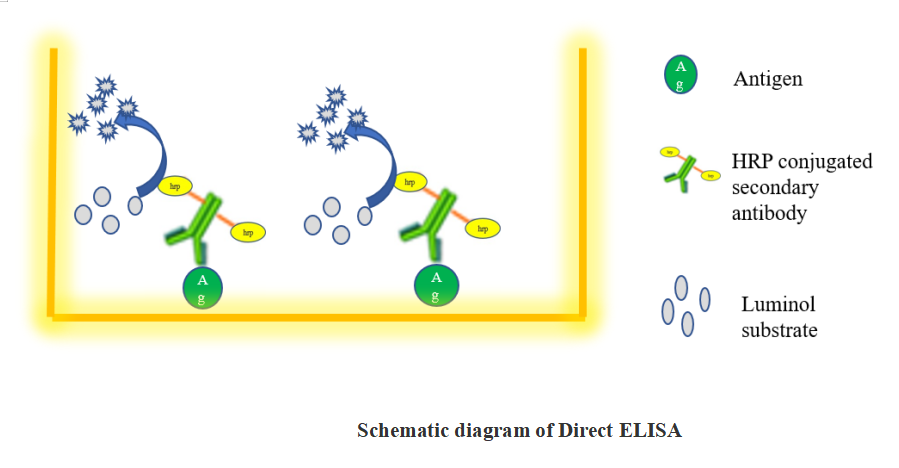
1.1 Direct ELISA
(1)Prepare antigen at 1 ug/mL in PBS and load 100 µL per well. Cover plate to minimize evaporation. Allow antigen to bind at 4° C overnight. The time required to bind antigen varies and may range from a few hours to overnight. Wash 3 times with wash buffer (PBS +0. 5%tween 20).
(2)Add 100 µL blocking solution (1% BSA in PBS) per well. Allow blocking to occur at 37ºC for 1 hours. Remove blocking solution and wash 3 times with wash buffer.
(3)Prepare enzyme conjugated secondary antibody by dilution in PBS or use the recommended dilution ranges. Load 100 µL of diluted enzyme conjugated antibody per well. Allow the conjugate to bind at 37ºC for 1 hours. Remove conjugate and wash 3 times with wash buffer.
(4)Add 100 µL of desired substrate solution (TMB) per well. Allow color to develop at room temperature and stop the reaction with stopping buffer (1M H2So4)
(5)Read absorbances at 450nm on plate using microplate reader.
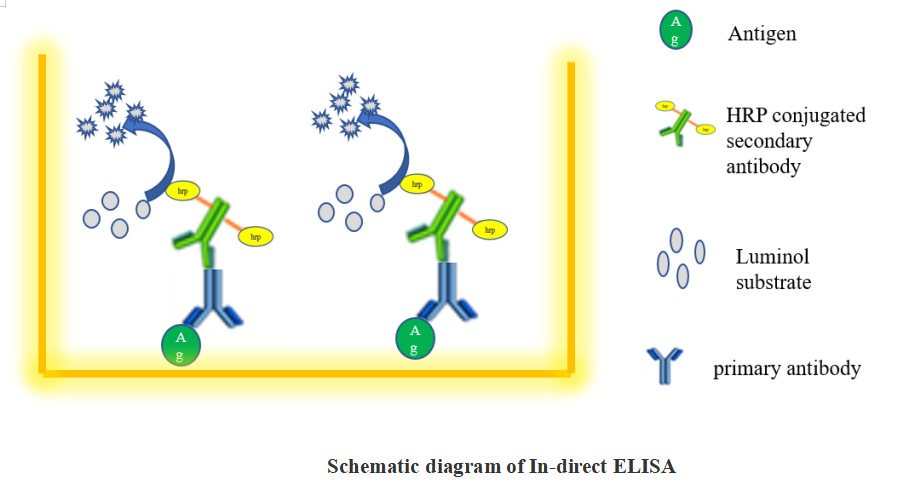
1.2 Indirect ELISA
An indirect ELISA is one where the primary antigen-specific antibody is recognized by a secondary conjugated antibody. The strength of this method is that different secondary antibodies can be used that recognize all antibody isotypes or specific isotypes (e.g., IgG).
Coating antigen to the microplate
(1)Coat the wells of a 96-well ELISA plate with purified antigen by pipetting 100 µL of purified protein antigen (1ug/mL in PBS) into each well of the plate.
(2)Cover the plate with an adhesive cover and incubate it overnight at 4°C to allow the antigen to bind to the plate.
(3)Upon incubation completion, remove the coating solution by flicking the plate over a sink.
Blocking
(1)Block the remaining protein-binding sites in the coated wells by adding 100 µL blocking buffer (1% BSA in PBS) per well.
(2)Incubate 1 hours at 37ºC.
(3)Following the incubation, remove the blocking buffer by flicking the plate and then wash plate with wash buffer.
Incubation with the primary antibody
(1)Prepare a serial dilution of the serum sample (which contains the primary antibody) or the primary antibody, to obtain a dilution range of 1 to 204,800, using 1X PBS.
(2)Add 100 µL of the serially-diluted serum samples to the wells.
(3)Cover plate with adhesive cover and incubate at 37ºC for 1 hours.
(4)Following the incubation, flick the plate over a sink and wash plate with wash buffer.
Incubation with the secondary antibody
(1)Add 100 µL of an enzyme-conjugated (HRP-conjugated) secondary antibody to each well.
(2)Incubate the plate for 1 hour at 37ºC.
(3)Following the incubation, flick the plate over a sink and then wash plate with PBS containing 1% Tween-20.
Detection
(1)Add 100 µL of the indicator substrate (3,3',5,5'-tetramethylbenzidine (TMB)) at a concentration of 1 mg/mL to each well.
(2)Incubate the plate with the substrate for 5-10 min at room temperature.
(3)After 10 min, stop the enzymatic reaction by adding 100 µL 1M Sulfuric acid (H2SO4).
Within 30 min of adding the stop solution, read the plate using a microplate reader at 450 nm to determine the absorbance of the wells.
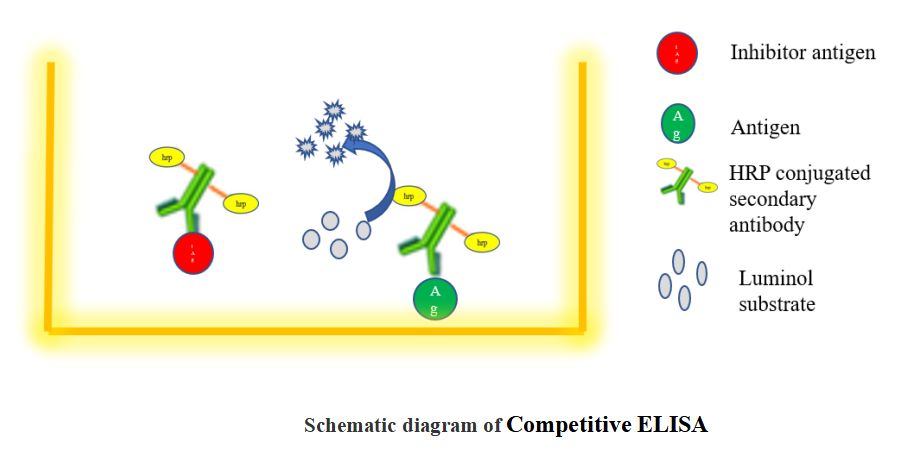
1.3 Competitive ELISA
The steps of a competitive ELISA are different from those used in indirect and sandwich ELISA, with the main difference being the competitive binding step between the sample antigen and the "add-in" antigen.
The sample antigen is incubated with the unlabeled primary antibody. These antibody-antigen complexes are then added to the ELISA plate, which has been pre-coated with the same antigen. After an incubation period, any unbound antibody is washed away. There is an inverse correlation between the amount of free antibody available to bind the antigen in the well and the amount of antigen in the original sample.
For example, a sample with abundant antigen would have more antigen-primary antibody complexes, leaving little unbound antibody to bind to the ELISA plate. An enzyme-conjugated secondary antibody specific to the primary antibody is then added to the wells, followed by the substrate.
Coating antigen to the microplate
(1)Coat the wells of a 96-well ELISA plate with 100 μL of purified antigen at a concentration of 1-10 μg/mL.
(2)Cover plate with an adhesive plate cover and incubate the plate overnight at 4°C.
(3)Following incubation, remove the unbound antigen solution from the wells by flicking the plate over a sink.
Blocking
(1)Block the remaining protein-binding sites in the coated wells by adding 200 μL of blocking buffer to each well, which can be either 5% non-fat dry milk or 1% BSA in PBS.
(2)Incubate the plate for 1 h at at 37°C.
Incubation sample (antigen) with the primary antibody
(1)While blocking the wells, prepare the antigen-antibody mixture by mixing 150 μL sample antigen and 150 μL of primary antibody for each well in the assay.
(2)Incubate this mixture for 1 h at 37°C.
Add antigen-antibody mixture to the well
(1)Now, remove the blocking buffer from the wells by flicking the plate over a sink.
(2)Then, wash the wells with 1X PBS containing 0.5% Tween-20 (wash buffer).
(3)Add 100 μL of the sample antigen-primary antibody mixture.
(4)Incubate the plate at 37°C for 1 h.
(5)Remove the sample mixture by flicking the plate over a sink.
(6)Then, wash the wells with wash buffer to remove any unbound antibody.
Add the secondary antibody
(1)Add 100 μL of an enzyme conjugated secondary antibody, which in this case is HRP-conjugated antibody, to each well.
(2)Incubate the plate for 1 h at 37°C.
(3)Following incubation, wash the plate with wash buffer.
Detection
(1)Add 100 μL of the substrate solution (TMB) to each well.
(2)Wait for 5-10 min.
(3)After the colour development, stop the enzymatic reaction by adding 100 μL 1M sulfuric acid to the wells. Then, measure the absorbance in a microplate reader within 30 min of adding the stop solution
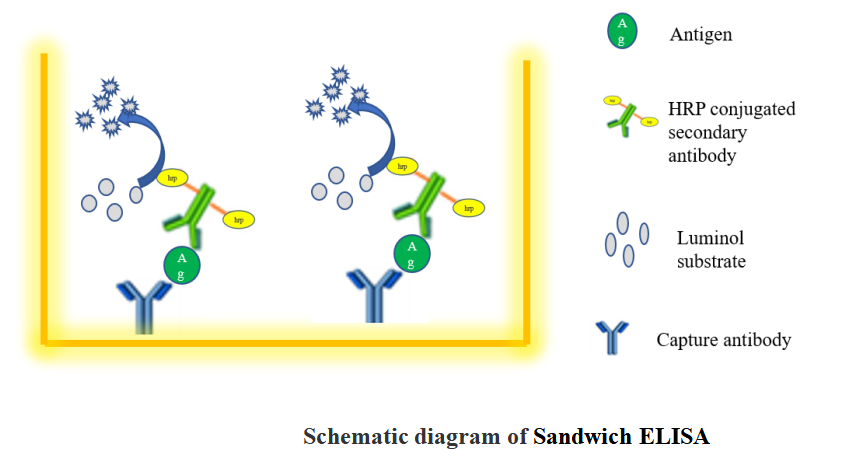
1.4 Sandwich ELISA
Introduction
A sandwich ELISA measures antigen between two layers of antibodies (capture and detection antibody). The target antigen must contain at least two antigenic sites capable of binding to antibodies.
Monoclonal or polyclonal antibodies can be used as the capture and detection antibodies in sandwich ELISA systems. Monoclonal antibodies recognize a single epitope that allows quantification of small differences in antigen.
A polyclonal is often used as the capture antibody to pull down as much of the antigen as possible. Sandwich ELISAs remove the sample purification step before analysis and enhance sensitivity (2–5 times more sensitive than direct or indirect).
Sandwich ELISA procedures can be difficult to optimize and tested match-paired antibodies should be used. This ensures the antibodies are detecting different epitopes on the target protein and do not interfere with the other antibody binding.
We are unable to guarantee our antibodies in sandwich ELISA unless they have been specifically tested.
Coating with capture antibody
(1)Coat the wells of a PVC microtiter plate with the 100ul of capture antibody at 1–10 μg/mL concentration in PBS.
(2) Unpurified antibodies (eg ascites fluid or antiserum or culture supernatant) may require increased concentration of the sample protein (try 10 μg/mL) to compensate for the lower concentration of specific antibody.
(3)Cover the plate with adhesive plastic and incubate overnight at 4°C.
(4)Remove the coating solution and wash the plate twice by filling the wells with 200 μL PBS containing 0.5% tween 20 (wash buffer).
Blocking and adding samples
(1)Block the remaining protein-binding sites in the coated wells by adding 200 μL blocking buffer (5% non-fat dry milk/PBS or 1% BSA in PBS) per well.
(2)Cover the plate with adhesive plastic and incubate for at least 1 h at 37°C.
(3)Wash the plate twice with 200 µL wash buffer.
(4)Add 100 μL of diluted samples to each well. Always compare signal of unknown samples against those of a standard curve. Run standards (duplicates or triplicates) and blank with each plate. Incubate for 1h at 37°C.
(5)Remove samples and wash the plate twice with 200 μL PBS.
Incubation with detection and secondary antibody
(1)Add 100 μL of diluted detection antibody to each well.
Check that the detection antibody recognizes a different epitope on the target protein to the capture antibody. This prevents interference with antibody binding. Use a tested matched pair whenever possible.
(2)Cover the plate with adhesive plastic and incubate for 1 h at 37°C.
(3)Wash the plate four times with wash buffer.
(4)Add 100 μL of conjugated secondary antibody diluted in PBS.
(5)Cover the plate with adhesive plastic and incubate for 1 h at 37°C.
(6)Wash the plate four times with PBS.
Detection
(1)Add 100 μL of the substrate solution (TMB) to each well.
(2)Wait for 5-10 min.
(3)After the colour development, stop the enzymatic reaction by adding 100 μL 1M sulfuric acid to the wells. Then, measure the absorbance in a microplate reader within 30 min of adding the stop solution
1.5 A summary of the advantages and disadvantages of the different ELISA techniques
|
ELISA |
Strengths |
Weaknesses |
|
Indirect |
1) High sensitivity due to the fact that multiple enzyme-conjugated secondary antibodies can bind to the primary antibody |
1) High background signal may occur because the coating of the antigen of interest to the plate is not specific (i.e., all proteins in the sample will coat the plate) |
|
2) Many different primary antibodies can be recognized by a single enzyme-conjugated secondary antibody giving the user the flexibility of using the same enzyme-conjugated secondary antibody in many different ELISA (regardless of the antigen being detected) |
||
|
3) Best choice when only a single antibody for the antigen of interest is available |
||
|
Sandwich |
1) The use of antigen-specific capture and detection monoclonal antibody increases the sensitivity and specificity of the assay (compared to the indirect ELISA) |
1) Optimizing the concentrations of the capture and detection monoclonal antibodies can be difficult (especially for non-commercial kits) |
|
2) Best choice for detecting a large protein with multiple epitopes (such as a cytokine) |
||
|
Competitive |
1) Impure samples can be used |
1) Requires a large amount of highly pure antigen to be used to coat plate |
|
2) Less sensitivity to reagent dilution effects |
||
|
3) Ideal for detecting small molecules (such as a hapten) |
|
 Guidence of GeneMedi's protocol / procedure for the diagnostics application:
Guidence of GeneMedi's protocol / procedure for the diagnostics application: 