


Immuno-oncology cell therapy
(CAR-T vs TCR-T vs CAR-NK vs ontolytic virus)
 Lentivirus Protocol Download
Lentivirus Protocol Download
In the past several decades, great progress has been made in the field of anti-tumor therapy, but there are also multiple strategies for tumors to evade the recognition and efficient suppression by the immune system. Therefore, a variety of immunotherapeutic targets (Fig. 1) and strategies have been developed to reactivate and reorganize the human immune system, such as CAR-T, TCR-T, CAR-NK, DC-CIK, oncolytic virus and so on.
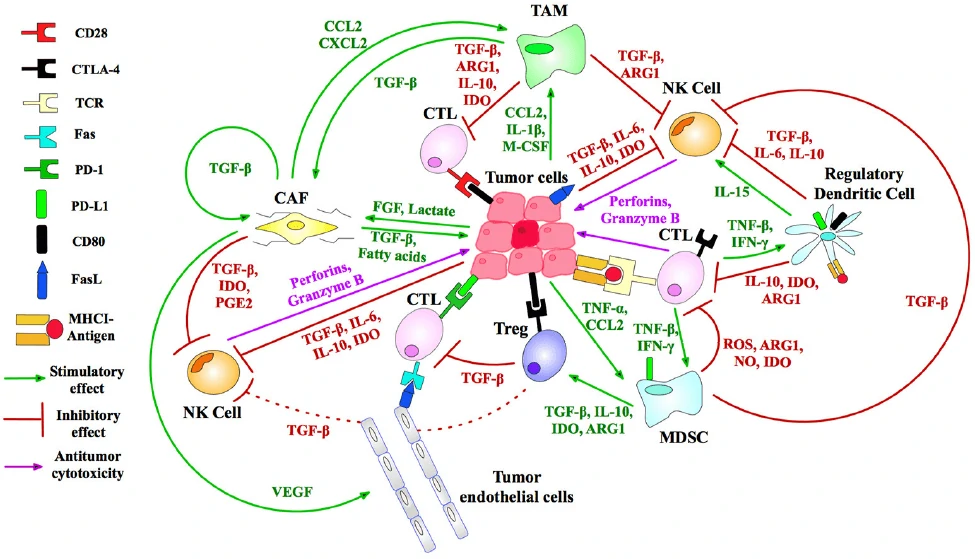 Figure 1 Immunosuppressive targets for tumor therapy [1].
Figure 1 Immunosuppressive targets for tumor therapy [1].CAR-T
Chimeric antigen receptor (CAR)-modified T cells (CAR-T) are T cells genetically engineered to express CAR (Fig. 2A) [2,3], which can specifically recognize their target antigen via the scFv binding domain of CAR, resulting in T cell activation to specifically target and destroy tumor cells [3-5]. To date, four generations of CAR have been developed according to the structure of the endodomain (Fig. 2B) [3]. Due to little ability to generate enough interleukin-2 (IL-2), the 1st generation CAR-T cells (such as Ag-specific CD3ζ (MFEζ)-CAR-T cells, alpha-folate receptor (FR) -CAR-T cells, CE7R-CAR-T cells, scFv(G250)-CAR-T cells, GD2- CAR-T cells and CD10- CAR-T cells) benefitted substantially from the combination of cytokines [6], and were used for the treatment of different tumors [7-10]. However, most of the studies using the 1st generation CAR-T cells did not show very satisfactory results because of the inadequate proliferation, cytotoxicity, and insufficient secreted cytokines in vivo. To overcome these shortcomings, the 2nd generation CAR-T cells were designed by adding intracellular signaling domains from various co-stimulatory protein receptors to the cytoplasmic tail of the CARs, [11-13]. The 2nd generation CAR-T cells, such as scFvCD19-CD137-CD3-CAR-T cells, MOv19-BBζ-CAR-T cells and scFvCD19-CD28-CD3ζ-CAR-T cells displayed better curative effects on B cell malignancies [12,14]. On the basis of the 2nd generation CAR-T cells, the 3rd generation CAR-T cells were produced with the addition of multiple signaling domains, such as CD3ζ-CD28-OX40 or CD3ζ-CD28-41BB, to promote cytokine production and killing ability. The 3rd generation CAR-T cells, such as CD20-CD28-CD137-CD3ζ-CAR-T cells and HER2-CAR-T cells were applied for the treatment of lymphoma and colon cancer, but with no better outcomes than the 2nd generation CAR-T cells, and the reasons need to be further studied [15,16]. Based on the 2nd generation CAR-T cells, the 4th generation CAR-T cells were obtained with the addition of IL-12, and can mediate T cell redirected for universal cytokine-mediated killing (TRUCKs). TRUCK T cells show great outcomes in disease therapy by augmenting T-cell activation, and also activating and recruiting innate immune cells to destroy the antigen-negative cancer cells in the targeted lesion, and can also be used for the therapy of viral infections, metabolic disorders and auto-immune diseases [17]. Overall, these successive generations of CAR-T cells have yielded remarkable efficacy in several types of cancer or tumor therapy, and some of them have been translated into clinical trials with few side effects (Table 1) [18].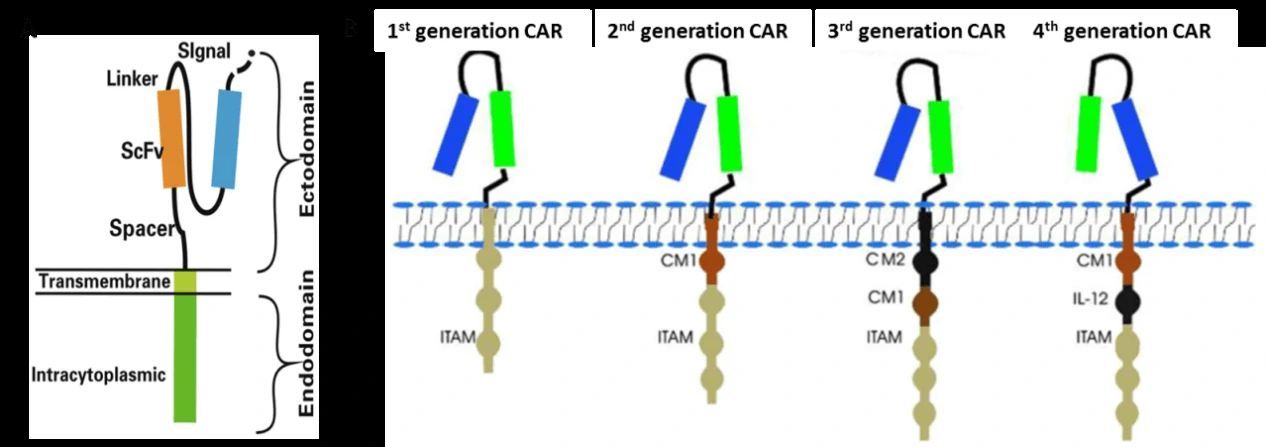 Figure 2 Structures of chimeric antigen receptor (CAR) [3]. (A) CAR structure: The CAR contains antigen recognition domain of thesingle-chain Fragment variant (scFv) derived from an antibody, transmembrane domain and an intracellular T cell activation domain of CD3ζ. (B) Evolution of CAR. ITAM: immunoreceptor tyrosine-based activation motifs. CM1: costimulatory molecule.
Figure 2 Structures of chimeric antigen receptor (CAR) [3]. (A) CAR structure: The CAR contains antigen recognition domain of thesingle-chain Fragment variant (scFv) derived from an antibody, transmembrane domain and an intracellular T cell activation domain of CD3ζ. (B) Evolution of CAR. ITAM: immunoreceptor tyrosine-based activation motifs. CM1: costimulatory molecule.| Antigen | Disease | In vitro, in vivo, in preclinical or in clinical trials | NCT/Reference |
| CD19 | haematologic malignancies | clinical trials | [19-22] |
| CD20 | haematologic malignancies | clinical trials | [23,24] |
| TRAIL receptor 1 | Lymphoma | In vitro | [25] |
| Kappa | Lymphoma | clinical trials | NCT00881920 |
| CD22 | follicular lymphoma, non-Hodgkin’s lymphoma | clinical trials | NCT02315612 |
| HA-1 H | Leukaemia | In vitro | [26] |
| NKG2D | Leukaemia | clinical trials | NCT02203825 |
| FAP | B cell chronic lymphocytic leukaemia | clinical trials | NCT01722149 |
| ROR1 | chronic lymphocytic leukaemia | clinical trials | NCT02194374 |
| CD138 | multiple myeloma | clinical trials | NCT01886976 |
| NY-ESO-1 | multiple myeloma | In vitro | [27] |
| Lewis Y | multiple myeloma | clinical trials | NCT01716364 |
| HER2 | Osteosarcoma | In vitro | [28] |
| HER2 | Breast cancer | In vitro | [29] |
| HER2 | Sarcoma | Clinical trial | NCT00902044 |
| HER2 | Metastatic cancer | Clinical trial | NCT00924287 |
| HER2 | Glioblastoma | Clinical trial | NCT01109095 |
| HER2 | Solid tumors | Clinical trial | NCT01935843 |
| CEA | Colorectal cancer | In vivo | [30] |
| CEA | Colorectal cancer | Clinical trial | NCT00673322 |
| CEA | Breast cancer | Clinical trial | NCT00673829 |
| CEA | Liver metastases | Clinical trial | NCT01373047 |
| CEA | Metastatic cancers | Clinical trial | NCT01723306 |
| CSPG4 | Melanoma, breast carcinoma | In vivo | [31] |
| EphA2 | Glioblastoma | In vivo | [32] |
| FR | Ovarian cancer | In vivo | [33] |
| IL-11Rα | Osteosarcoma | In vivo | [34] |
| IL-13Rα2 | Glioblastoma | Preclinical trial | [35] |
| IL-13Rα2 | Malignant glioma | Clinical trial | NCT02208362 |
| IL-13R | Glioma | Preclinical trial | [36] |
| CD171 | Neuroblastoma | Clinical trial | NCT02311621 |
| EGFR | Advanced EGFR-positive solid tumors | Clinical trial | NCT01869166 |
| EGFR | Advanced glioma | Clinical trial | NCT02331693 |
TCR-T
T-cell receptor (TCR)–engineered T-cell therapy (TCR-T) is one potentially powerful treatment that utilizes genetically modified natural T cells to specifically target tumors and destroy tumors with greater potentials [37]. Unlike CARs, TCRs rely on their interactions with peptide-major histocompatibility complex (pMHC), formed by peptide [38,39], generated from intracellular antigen proteolysis, bound with MHC (Fig. 3) [40]. Besides TCR, additional costimulatory or co-inhibitory signals are also required for TCR-T function. For example, CD4 on the surface of helper T cells, which binds to class II MHC complex, and CD8 on the surface of cytotoxic T cells, which binds to class I MHC complex, activates the TCR-T mediated cell destruction [41], while cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) are responsible for T cell signaling extinguisher [42]. Given that only ~28% antigens are expressed on the cell surface, it is difficult for most of the antigens to be recognized by CAR [43]. Therefore, compared to CAR-T, TCR-T cell therapy shows remarkable advantages over the destruction of tumor cells with intracellular antigens [44]. To date, TCR-T mediated cell therapy has achieved many promising curative potency in both solid tumors and hematological cancers, and some of them have been applied in clinic trials (Table 2).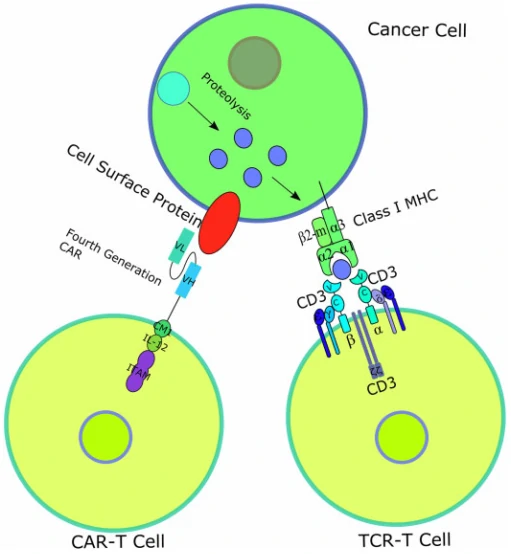 Figure 3 Diagrams of CAR-T and TCR-T-cell therapy [45]. Unlike CAR, TCR is a heterodimer containing 2 different transmembrane polypeptide chains: α chain and β chain. The α chain and β chain each consist of a constant region and a variable region, the former anchoring the chain inside the T-cell surface membrane and the latter recognizing and binding to the antigen presented by MHCs [46].
Figure 3 Diagrams of CAR-T and TCR-T-cell therapy [45]. Unlike CAR, TCR is a heterodimer containing 2 different transmembrane polypeptide chains: α chain and β chain. The α chain and β chain each consist of a constant region and a variable region, the former anchoring the chain inside the T-cell surface membrane and the latter recognizing and binding to the antigen presented by MHCs [46].| Antigen | Disease | Phase of clinical trials | NCT/Reference |
| PRAME | Unknown | Phase I/II | NCT03503968 |
| PRAME | Acute myeloid leukemia, myelodysplastic syndrome | Phase I | NCT02743611 |
| MAGE-A3/A6 | Unknown | Phase I | NCT03139370 |
| Unknown | Head and neck squamous cell carcinoma, squamous cell NSCLC | Phase I | NCT03139370 |
| MAGE-A10 | Non-small cell lung carcinoma | Phase I | NCT02592577 |
| NY-ESO-1 | Synovial sarcoma | Phase I/II | NCT01343043 |
| NY-ESO-1 | Multiple myeloma | Phase I/II | NCT01892293 |
| NY-ESO-1 | Multiple myeloma | Phase I/II | NCT01352286 |
| NY-ESO-1 | Ovarian cancer | Phase I/II | NCT01567891 |
| NY-ESO-1 | Melanoma | Phase I/II | NCT01350401 |
| NY-ESO-1 | Non-small cell lung carcinoma | Phase I/II | NCT02588612 |
| AFP | Hepatocellular cancer | Phase I | NCT03132792 |
| NY-ESO-1 | Refractory multiple myeloma | Phase I | NCT03168438 |
| MAGE-A10 | Urinary bladder cancer, head and neck cancer, melanoma | Phase I | NCT02989064 |
CAR-T vs TCR-T CAR-T and TCR-T both are gene-engineered technologies targeted for the destruction of tumors by improving the recognition and destroy potentials of T cells, thus called “T cell receptor redirection” technologies. However, there are also some differences in the two technologies: 1) CAR-T recognizes their target antigen via the scFv binding domain of CAR, while TCR-T relies on the interactions with peptide-major histocompatibility complex (pMHC) and requires co-stimulatory od co-inhibitory signals; 2) CAR-T recognizes and targets cell surface antigens, while TCR-T can target both cell surface antigens and intracellular antigens; 3) CAR-T shows great therapy outcomes in the treatment of hematological cancers but little effects on solid tumors, while TCR-T displays encouraging curative outcomes in the therapy of solid tumors.
CAR-NK
Although CAR-T has been applied in clinic, quite a lot of side effects, such as off-target, cytokine release syndrome (CRS) etc., pose a great restriction in their further clinical application [47]. Based on natural killer (NK) cells, CAR-NK cell therapy is much safer than CAR-T therapy and becomes a good candidate for targeting and combating solid tumors [48,49]. Based on the four generation of CAR, CARs in CAR-NK are designed with the addition of personalized proteins, such as DNAX-activation protein (DAP) 12 and NKG2 member D (NKG2D), which show great anti-tumor activity in acute myeloid leukemia (ALL), osteosarcoma, and prostate tumors [50-52]. Therefore, CAR-NK cells not only have the ability of CAR to specifically recognize antigen-expressing tumors, but also can destroy tumors via NK cell receptors. To date, numerous CAR-NK cell therapies have been used for anti-tumor therapy in clinical trials (Table 3).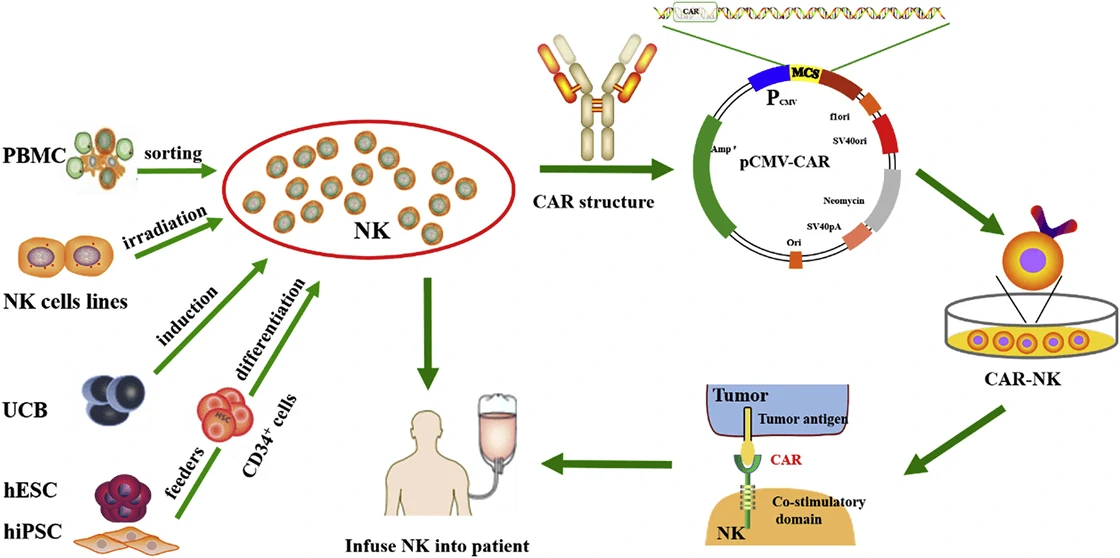 Figure 4 Schematic of CAR-NK cell therapy [53]. Gene engineered natural killer (NK) cells are infused into patients to destroy tumor cells. PBMC, peripheral blood mononuclear cell; UCB, umbilical cord blood; ESCs, human embryonic stem cells; iPSCs, human induced pluripotent stem cells.
Figure 4 Schematic of CAR-NK cell therapy [53]. Gene engineered natural killer (NK) cells are infused into patients to destroy tumor cells. PBMC, peripheral blood mononuclear cell; UCB, umbilical cord blood; ESCs, human embryonic stem cells; iPSCs, human induced pluripotent stem cells.
| Antigen | Disease | NK source | Phase of clinical trials | NCT/Reference |
| CD7 | Lymphoma,leukemia, | NK-92 | Phase I/II | NCT02742727 |
| CD19 | Lymphoma,leukemia, | NK-92 | Phase I/II | NCT02892695 |
| CD33 | Acute myeloid leukemia | NK-92 | Phase I/II (complete) | NCT02944162 |
| MUC1 | Solid tumors | NK-92 | Phase I/II | NCT02839954 |
| NR | Non-small cell lung carcinoma | NK-92 | Phase I | NCT03656705 |
| HER2 | Glioblastoma multiforme | NK-92 | Phase I | NCT03383978 |
| CD19 | B-acute lymphocytic leukaemia | PB-NK | Phase II | NCT01974479 |
| CD19 | B-acute lymphocytic leukaemia | PB-NK | Phase I (complete) | NCT00995137 |
| CD19 | B-lymphoma | UCB-NK | Phase I/II | NCT03056339 |
Viral vectors used in CAR-T, TCR-T and CAR-NK
The broad application of gene engineered adoptive cell therapy, such as CAR-T, TCR-T and CAR-NK, in clinical trials for the therapy of cancers led to the development of safety-enhanced self-inactivating (SIN) vectors, such as γ-retroviral (gRV), lentiviral (LV) vectors, adenovirus, and AAV vectors. Scalable manufacturing processes for the production of gRV vectors (Fig. 5) and LV vectors using transfection in closed-system bioreactors in compliance with current good manufacturing practices (cGMP) for clinical applications have been developed and optimized [54,55].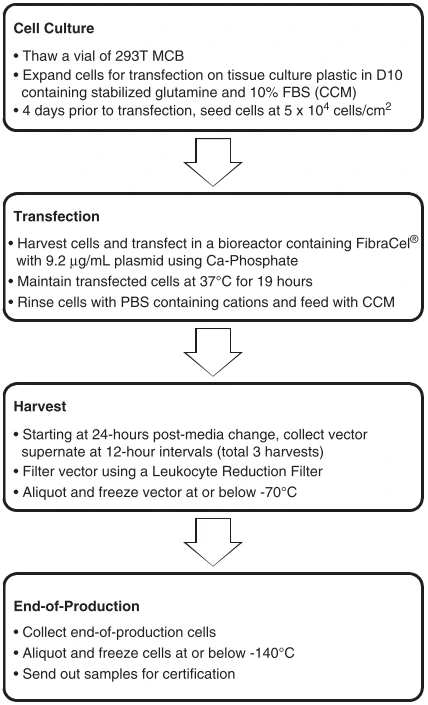 Figure 5 Clinical manufacturing process flow chart of γ-retroviral vectors based on adherent cells [55].
Figure 5 Clinical manufacturing process flow chart of γ-retroviral vectors based on adherent cells [55].DC-CIK
Cytokine-induced killer cells (CD3+ CD56+ cells, CIK cells) [56] are non-major histocompatibility complex-restricted natural killer T lymphocytes and exhibit stronger anti-tumor and cytolytic activities than lymphokine-activated killer cells [57-59]. Co-cultured with dendritic cells (DCs) or Ag-DC cells, the selective cytotoxicity and anti-tumor immunity of CIK cells would be significantly enhanced, i.e. DC-CIK therapy [60,61]. Compared with traditional treatment, DC-CIK immunotherapy not only improves the clinical indices, but also shows mild adverse and reduces mortality [62].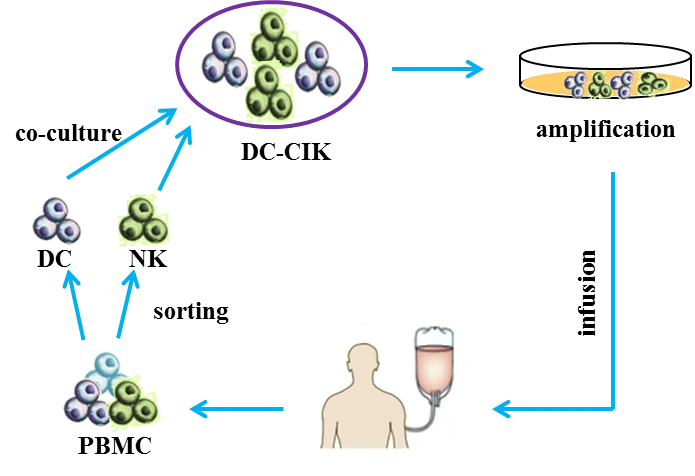 Figure 6 Schematic of DC-CIK cell therapy [63]. Co-cultured dendritic cells (DCs) and cytokine-induced killer cells (CIKs) (DC-CIK) are infused into patients for anti-tumor therapy.
Figure 6 Schematic of DC-CIK cell therapy [63]. Co-cultured dendritic cells (DCs) and cytokine-induced killer cells (CIKs) (DC-CIK) are infused into patients for anti-tumor therapy.Oncolytic virus
Oncolytic virotherapy is a novel therapeutic modality utilizing oncolytic viruses (such as herpes simplex-, pox-, parvo-, or adenoviruses), which selectively replicate in cancer cells, to directly induce tumor cell lysis or indirectly reactivate human immune system to mediate tumor destruction (Fig. 7) [64]. Through decades’ optimization, a great number of oncolytic virus therapies have been put into clinical trials, such as ONYX-015 (E1B-deleted adenovirus) [65], DNX-2401 (Ad5) [66], CG0070 (Ad5 carrying GM-CSF gene) [67], OBP-301 (Ad5-hTERT-E1A/B) [68], G207 (a conditionally-replicative HSV-1 with ICP34.5 deletion and UL39 disruption) [69], and JX-594 (Pexa-Vec, GM-CSF-enhanced vaccinia virus with TK gene disruption) [67]. Among them, Amgen's T-VEC (a modified HSV-1 with expression of GM-CSF but deletions in ICP34.5 and ICP47 genes) was the first oncolytic virus approved by America Food and Drug Administration (FDA) for the treatment of melanoma in 2015 [70]. Moreover, when combined with the other therapy, such as Anti-PD-1 immunotherapy, oncolytic virus can significantly promote immune recognition of cancer cell as well as T cell infiltration, and lead to high response rate in patients with advanced melanoma [71].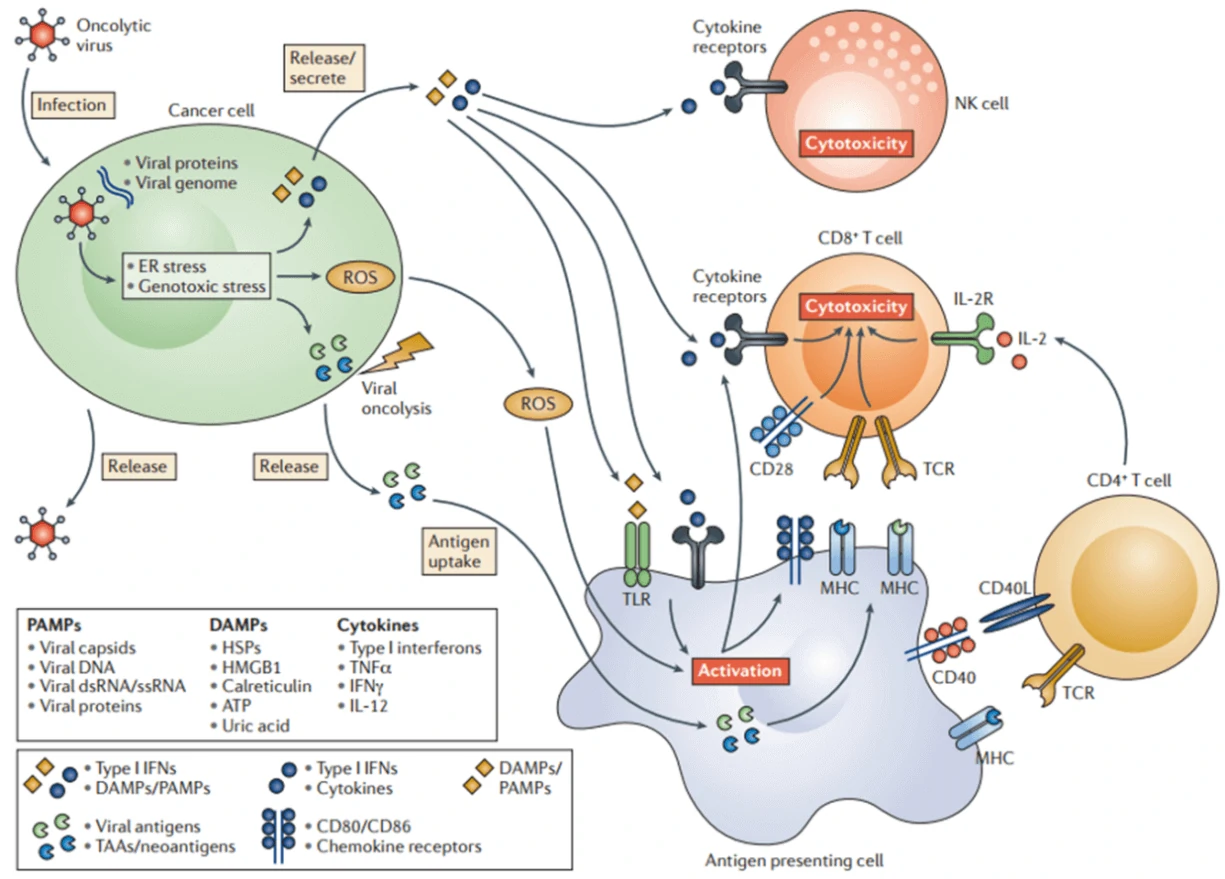 Figure 7 Oncolytic viruses-induced anti-tumor immunity [64]. Oncolytic viruses mediate direct cancer lysis and indirect activation of anti-tumor immune responses.
Figure 7 Oncolytic viruses-induced anti-tumor immunity [64]. Oncolytic viruses mediate direct cancer lysis and indirect activation of anti-tumor immune responses.| Oncolytic viruses | Disease | Description | NCT/Reference |
| Adenovirus | Hepatic carcinoma | Golgi protein 73 (GP73) needed | [72] |
| Adenovirus | Pancreatic carcinoma, prostate cancer | Tat-PTD modified hexon and Ad5/35 | [73] |
| Adenovirus | Gastric cancer | Acetylcholinesterase (AChE) | [74] |
| Adenovirus | Gastric cancer | OBP-301, telomerase-specific | [75] |
| Herpes simplex virus 2 | Colon cancer | No virus modification or co-therapies | [76] |
| Vaccinia | Colon cancer | Viral Thymidine kinase (TK) deficiency | [77] |
| Measles virus | Hepatocellular carcinoma, colon cancer | Retargeted to CD133 | [78] |
| Herpes simplex virus | Ovarian cancer | Interleukin−12 | [79] |
| ONYX-015 (adenovirus) | Malignant mesothelioma cells | EB1 gene deletion | [65] |
| DNX-2401 (adenovirus) | Glioblastoma and gliosarcoma | NCT02197169 | |
| CG0070 (adenovirus) | Bladder cancer | carrying CM-CSF gene | NCT02143804, NCT02365818 |
| OBP-301 (adenovirus) | carrying hTERT-E1A/B | NCT0229385 | |
| HF10 (Herpes simplex virus) | Solid superficial malignant tumors | deletions in neurolatency genes UL43, UL49.5, UL55, and UL56 | NCT02428036 |
| G207 (Herpes simplex virus) | Glioblastoma | a conditionally-replicative HSV-1 with ICP34.5 deletion and UL39 disruption | NCT00157703 |
| JX-594 (Vaccinia Virus) | Hepatocellular Carcinoma, colorectal cancer | GM-CSF-enhanced vaccinia virus with TK gene disruption | NCT02562755 |
| GL-ONC1 (Vaccinia Virus) | Head and neck cancer | TK-inactivated | NCT01443260 |
| Reolysin (Retrovirus) | Metastatic melanoma or pancreatic cancer | NCT02514382, NCT02444546 | |
| Cavatak™ (Coxsackievirus A21) | Melanoma | Overexpress ICAM-1 and DAF | NCT02565992, NCT02316171 |
Taken together, different kinds of immune-oncology cell therapy show great variety in their curative effects. Although promising therapeutic outcomes in acute lymphocytic leukemia (ALL) and large B cell lymphomas have been achieved by CAR-T [80,81], little therapeutic effects were acquired in solid tumors as well as tumors with intracellular antigens, and multiple side effects, such as cytokine release syndrome, prohibited the broad application of CAR-T [82-84]. TCR-T displays better effects for the treatment of hematopoietic cancers and solid tumors, but requires MHC and co-stimulatory or co-inhibitory signals to combat tumors [45]. Without the drawback of CAR-T, CAR-NK is safer than CAR-T and presents excellent tumor elimination ability in both hematopoietic cancers and solid tumors via CAR-dependent and NK receptor-dependent mechanisms [53]. DC-CIK immunotherapy shows improved immunologic function, reduced mortality, and mild adverse effects in both hematologic malignancies and solid tumors, and may be suitable for patients that are intolerance to radiotherapy and chemotherapy [62,85]. Without the need of defined antigens included in the vectors, oncolytic viruses show a durable anti-tumor effects by directly infecting and lysing tumor cells in situ and also activating the host immune response to eliminate tumor cells, thus displaying curative effects on both hematopoietic cancers [86,87] and solid tumors [64]. Moreover, these immune-oncology cell therapies can also be combined with programmed cell death protein 1 (PD-1) antibody or PD-1 inhibitor, showing reduced drug toxicity and enhanced the tumor cytotoxicity [88]. For example, CD19-CAR-T cell carrying the single-chain variable fragment (scFv) of antibody against PD-1 exhibited potent therapeutic effects superior to those of conventional CAR-T cells [89]; oncolytic Herpes Simplex Virus Type 2 encoding an antibody against PD-1 presented an enhanced therapeutic efficacy with a durable antitumor response both in the tumor microenvironment and in the systemic immune system [90].
View Knowledge Base - Landscape of Advanced cell therapy>>
AAV(Adeno-Associated Virus) vector system
AAV Rep-Cap plasmids (serotypes-specific AAV RC plasmids)
